《表1 中日细胞治疗产品监管法规对比》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
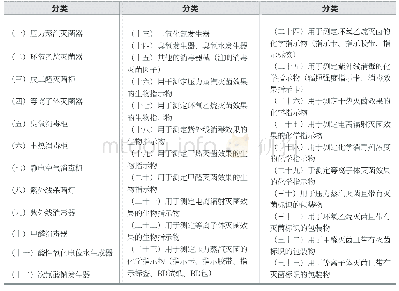
2017年12月,原国家食品药品监督管理总局(CFDA)颁布了《细胞治疗产品研究与评价技术指导原则(试行)》并开始实施。该文件为细胞治疗药品的研发、生产与注册指明了路径,旨在进一步规范细胞治疗产品的研发,提高其安全性、有效性和质量可控性水平。关于临床试验,其中指明:鉴于细胞治疗产品特殊的生物学特性,在临床试验中,需要采取不同于其他药物的临床试验整体策略。而在干细胞临床研究管理方面,2016年原CFDA发布《干细胞临床研究管理办法(试行)》,其中对干细胞临床研究过程及干细胞制剂等作了详细的要求和规范,同时指出干细胞治疗相关技术将不再按照第三类医疗技术进行管理[12]。随即原国家卫生计生委与原CFDA共同成立了国家干细胞临床研究专家委员会,其成员由干细胞基础及临床相关专业、干细胞制剂制备和质量控制等领域33位专家组成,为干细胞临床研究规范管理提供技术支撑。2019年国家卫健委发布了《体细胞治疗临床研究和转化应用管理办法(试行)(征求意见稿)》和《生物医学新技术临床应用管理条例(征求意见稿)》,这2份监管文件主要关注的焦点都是细胞治疗,其明确了医疗机构作为责任主体,可以进行体细胞治疗等新技术的临床研究,在获得安全有效性数据后,可以申请临床应用并收费。为了弥补我国对细胞治疗产品生产和质量检查在法规层面和技术层面的空白,进一步完善细胞治疗产品监管长效机制,国家疫苗检查中心暨国家药品监督管理局(NMPA)食品药品审核查验中心于2019年11月28日发布了《GMP附录-细胞治疗产品》(征求意见稿),适用于从供体材料的运输、接收、产品生产和检验到成品放行、储存和运输的全过程(见表1)。
| 图表编号 | XD00141128900 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.12.25 |
| 作者 | 雕钰惟、梁毅 |
| 绘制单位 | 中国药科大学国际医药商学院、中国药科大学国际医药商学院 |
| 更多格式 | 高清、无水印(增值服务) |
![表5 中日西式香肠产品标准指标设立对比[10-11,16-17]](http://bookimg.mtoou.info/tubiao/gif/RLYJ202009013_03900.gif)
![表6 中日西式香肠产品标准关键指标对比[10-11,16-17,19-20]](http://bookimg.mtoou.info/tubiao/gif/RLYJ202009013_04400.gif)



{kind=link}