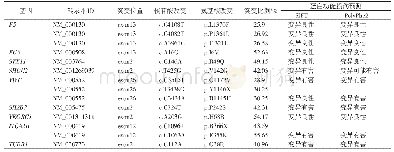
《表4 外显子测序Idels变异数据Tab.4 Idels mutation data of exon sequencing》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
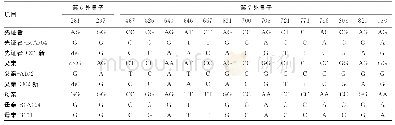
通过比对,得到大量的SNP(表3)和Indel位点(表4)之后,通过以下几步筛选,来获取疾病的候选致病基因位点:首先,去除不影响基因功能的同义突变,保留影响基因功能的非同义突变和剪接位点;其次,通过数据库过滤,去除dbSNP等遗传变异数据库中的普通变异和正常对照中的变异;再次,隐马尔可夫模型预测(Hidden Markov Model,HMM),该模型是在信息分析的流程中用到的一种数学模型,其原理是根据家系内患者间的基因型表现及上下文(指该SNP位点两侧位点的基因型)推算2个患者当前位点所在片段是否来自相同祖先,如果“是”则有致病的可能。随后过滤家系内正常对照样本,与病例组共有的突变筛选(患者之间取交集);最后,通过查阅基因功能与表型的相关性分析确认候选致病基因。因为同义变化不太可能致病,我们将同义变化筛选出来,基于这一假设,该家系疾病相关的基因应当不存在于一般人群[8]。我们进一步假设2个患者进行外显子测序后应表现为同样的NS/SS插入或缺失变异,那么此插入或缺失变异即有可能是导致此家系成员患病的致病基因。但是家系内每个成员的插入或缺失变异的数量非常大,其共同的变异位点也可能是多个,这种情况下增加测序标本的数量,从3个或4个家系成员标本中找出其共同变异就可以缩小研究范围。因此,在本研究中找寻4名患者共同的外显子基因变异位点,即为可能的致病基因变异。
| 图表编号 | XD0043930500 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.01.05 |
| 作者 | 孙甫、杨姣、郭发刚、袁启令、刘亮、戴兰兰、张刘军、张银刚 |
| 绘制单位 | 西安医学院第一附属医院骨科、西安医学院全科医学系、西安医学院全科医学系、西安交通大学医学院第一附属医院骨科、西安交通大学医学院第一附属医院骨科、深圳华大基因、深圳华大基因、西安交通大学医学院第一附属医院骨科 |
| 更多格式 | 高清、无水印(增值服务) |




{kind=link}