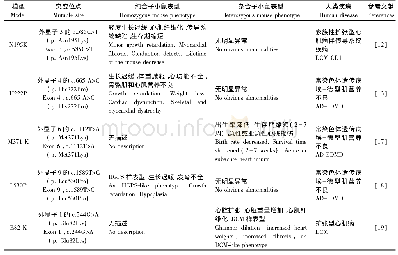
《表1 TSHR基因错义突变位点》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
本征是常染色体隐性遗传性疾病,从理论上讲,TSH与TSH受体作用过程中的任何环节成受体后障碍均可引起,常见的病因有:1)TSH-β亚基基因突变[6];2) TSH受体基因突变[7];3) Gsa亚基基因突变[8];4) TSH受体信号转导途径的先天性缺陷[9];5) TSH受体后缺陷等[9]。Gsa亚基基因突变,可出现激素特异性丢失现象。TSH受体基因突变致HIS,相对研究的较多,其遗传方式符合常染色体隐性遗传的特点[4]。TSHR基因突变可引起MIS的证据,最先由Gu等于1995年,在对遗传性甲低鼠(hyt/hyt)的研究,此鼠先天性甲状腺功能低下的发生,是由于TSHR基因突变,其突变的位点在鼠TSHR基因556位密码子的错义突变(Pro556leu))所致[10]。后自90年代中期,许多学者对人类有TSH不反应的甲减患者进行基因分析,之后陆续发现具有TSHR基因突变的先天性甲状腺功能低下家系,证实了TSHR基因的存在,而且突变的位置、类型等均不一致,目前已见报道的TSHR基因突变类型有:1)错义突变:是由于单个碱基的替代,三联密码的碱基组或发生变化,使原来的氨基酸密码子被另一氨基酸的密码子所替代,此类型突变是最常见的类型,所涉及的类型见表1[11]。2)无义突变:是由于编码密码子发生碱基替换,致该密码子突变为终止密码而使该肽键合成提前终止,而形成截断的TSH受体,如:1)第324位密码子[12],由于C→T转换,使TSHR基因的合成提前在第324位截断而形成比正常少440个氨基酸的异常多肽链;2)第546位密码子[12,15]:由于在该密码子第二位碱基发生G→A转换,使之成为终止密码子TAG,合成在546位提前截断,结果比正常少218个氨基酸;3)缺失/插入突变:目前仅在一个家系中检出[13],缺失位点在1217~1274处,缺失了18bp,同时又另有4bp插入,即在第419促氨基酸后提前出现了终止密码,提前在第420位合成截断,缺少了344个氨基酸。该综合征发病的基因型,通常的常染色体隐性遗传病,是纯合子发病,而TSHR基因突变发病的基因型,大多数为双重杂合子。亦有单个位点的纯合子突变,而且大部分突变位于TSHR基因的穿膜区,即第10外显子编码区,胞位区的第1、4、6外显子序列较短,故突变位点较少,突变基因型与临床表现型存在异质性[16]。第10外显子对甲状腺发育、功能调节比较重要[14]。R450H型突变首先由Nagashima等[17]于2001年报道2位TSH抵抗的2位兄弟的TSHR基因,结果C位的突变基因型一致,均为R450H和G4983双位点杂合子型突变,血清TSH升高,仅有轻度甲状腺发育不良,其父母为1个位点突变杂合子,但均未发病。
| 图表编号 | XD00204783700 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2021.02.20 |
| 作者 | 谢辉、刘国良 |
| 绘制单位 | 沈阳市红十字会医院、中国医科大学第一附属医院 |
| 更多格式 | 高清、无水印(增值服务) |

{kind=link}