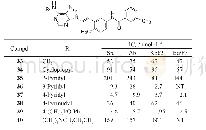
《Table 3 Structures and activity of varied C6 substituent.NT=not test》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
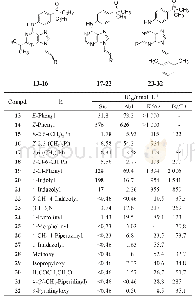
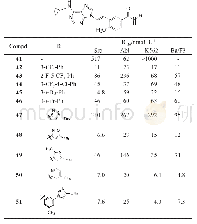
合成了上述对接设计的化合物33(表3),活性评价结果证实对Src和Abl都有较高活性,而且抑制白血病细胞活性也达到亚纳摩尔水平,而且在10μmol·L-1浓度下对正常细胞无作用,提示有高选择性。大鼠药代动力学实验表明33的口服利用度(F)20%,半衰期很短,是由于C6的甲胺基发生氧化去甲基作用。故而合成了环丙基化合物34(环丙基代谢稳定性高于甲基),仍保持了酶和细胞活性,而改善了药代,F=40%,血浆中有高暴露量。环丙基的亲脂性强于甲基,推论进一步增高亲脂性会有利于活性。为此合成了芳环35~40,吡啶和嘧啶化合物活性都很高,但2-吡啶基化合物35却很低,可能是由于2'-N与嘌呤N1的电性排斥作用,芳环扭转,破坏了共面性,而36和37没有排斥作用(但嘧啶也有邻位排斥作用,活性下降却不明显)。化合物39移植了9的模块,酶和细胞活性特别高。40的活性很弱。然而这些高活性化合物的大鼠药代都不好,因而将C6-环丙胺基固定做进一步优化。
| 图表编号 | XD0061345200 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.05.12 |
| 作者 | 郭宗儒 |
| 绘制单位 | 中国医学科学院,北京协和医学院药物研究所 |
| 更多格式 | 高清、无水印(增值服务) |




{kind=link}