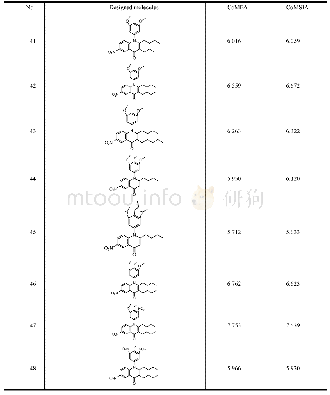
《Table 3.Structures and Predicted Activities of the Designed Molecules》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
本系列图表出处文件名:随高清版一同展现
《3D-QSAR and Surflex Docking Studies of a Series of Alkaline Phosphatase Inhibitors》
The CoMFA and CoMSIA models of different field contribution analysis maps provided important information for modifying the structural features.Compound 19,which has the most excellent potency,was chosen as template for designing new molecules.A large green polyhedral area surrounded rings A and C.Therefore,the butyl of ring A was retained.As shown in docking study,the oxygen atoms of rings A and C form three hydrogen bonds with TNAP protein,so those oxygens were retained.The new putative active compounds designed by modifying compound 19 at the 3-posi-tion of ring A and 2,3,4-position of ring C are shown in Table 3.In addition,the activities of the designed compounds were evaluated using the CoMFA and CoMSIA models established above.Most of the new compounds(No.41~48)were predicted with better activity than compound 19,with the exception of molecule 45,presumably because the propyl is too bulky at the 3-position of ring C.
| 图表编号 | XD0034853000 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.01.01 |
| 作者 | 舒茂、武涛、王必武、李静、徐春媚、林治华 |
| 绘制单位 | School of Pharmacy and Bioengineering,Chongqing University of Technology、School of Pharmacy and Bioengineering,Chongqing University of Technology、School of Pharmacy and Bioengineering,Chongqing University of Technology、School of Pharmacy and Bioengineerin |
| 更多格式 | 高清、无水印(增值服务) |




{kind=link}