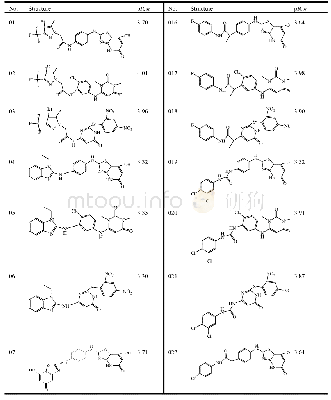
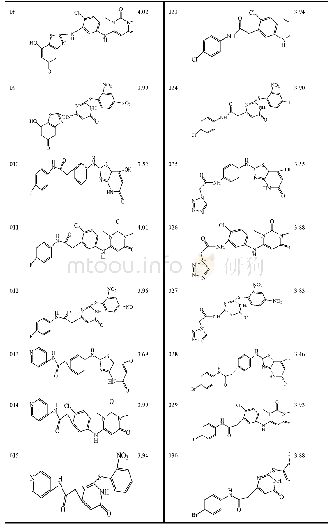
《Table 2.Structures and Predicted p IC50 Values of New Designed Molecules》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
本系列图表出处文件名:随高清版一同展现
《Comprehensive 3D-QSAR and Binding Mode of DAPY Inhibitors Using R-group Search and Molecular Docking》
We used the screened R-groups to construct new molecules with higher inhibitory activity.The contribution values of R-groups were determined according to the scores predicted by the Topomer CoMFA model.In general,R groups with higher priority contribution values were used to replace the R group of template molecule in the same limit of TOPDIST.Different molecules with specific activity were obtained according to the contribution values of the R groups and the structure of the template compound.Thus,the new molecules have similar structures to that of the known training compounds,in order to guarantee the category consistency between the newly designed molecules and the training compounds,and have different R-groups to ensure the improvement of bioactivity of inhibitors.In this work,we virtually screened R1 and R2 groups in the 130,000 drug-like molecules of the ZINC database,respectively.We treated compound 3 of the training set as a template to extract the top 10 R1-and3 R2-groups.The structures and activities of the 30new compounds are displayed in Table 2.It can be seen from Table 2 that the activities of 30 new compounds are higher than that of the training set molecules.And as shown in Table 2,new compounds have higher activities because there is bulky substituent at the Ra-position of molecules ring.This is consistent with the analysis of 3D contour of Topomer CoMFA model.And the structures of the newly designed molecules were optimized by adding hydrogen and suitable electric charge in consistency with what was described previously in establishing the Topomer CoMFA model section.The p IC50 value for each new molecule was predicted by the QSAR model[33,34].Compared to the data set molecules,we can easily find that newly designed molecules still have aromaticity and conform to the interpretation of the 3D coutour,although the structure is slightly different.These advantages of new molecules provide theoretical guidance for the discovery of new drugs.
| 图表编号 | XD0034853400 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.01.01 |
| 作者 | 仝建波、王洋、雷珊、秦尚尚 |
| 绘制单位 | College of Chemistry and Chemical Engineering,Shaanxi University of Science and Technology、Shaanxi Key Laboratory of Chemical Additives for Industry、College of Chemistry and Chemical Engineering,Shaanxi University of Science and Technology、Shaanxi Key Lab |
| 更多格式 | 高清、无水印(增值服务) |



{kind=link}