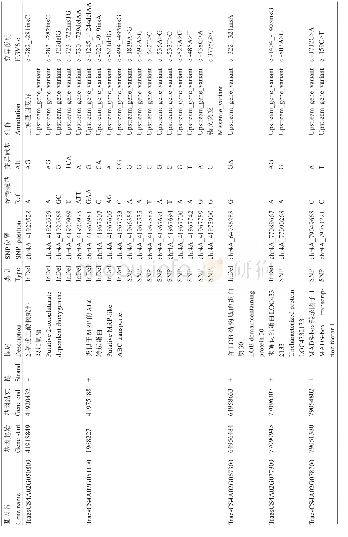
《表2 全基因组测序发现的胆汁酸代谢相关基因的变异类型》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
本系列图表出处文件名:随高清版一同展现
《一例钠牛磺胆酸共转运多肽缺陷病患儿临床和遗传学分析》
注:[AD]常染色体显性遗传;[AR]常染色体隐性遗传;[DR]双基因隐性遗传;[-]未知;*表示已报道。
全基因组测序未发现影响胆汁酸稳态的其它50种遗传病致病基因的插入、缺失、经典剪接位点变异等致病性变异,虽然SLCO1B1基因检出1个无义突变,AKR1D1和CFTR基因检出3个错义突变,然而其相关疾病在临床表型、遗传方式等方面均与本患儿临床和实验室表现不相符,见表2。进一步对3个错义突变采用Poly Phen-2、Mutation Taster和SIFT软件进行致病性预测,其中AKR1D1基因的c.797G>A(p.R266Q)变异虽三个软件的预测结果分别为可能致病、致病和影响蛋白质功能,但其表型为先天性胆汁酸合成障碍2型,主要表现为胆汁淤积性黄疸,脂肪、脂溶性维生素吸收障碍,血清转氨酶升高,但血清总胆汁酸正常甚至降低,而γ-谷氨酰转肽酶水平不高,与本患儿的临床和生化表型特征不符;其余2个错义变异软件预测结果均无致病性且变异位点的氨基酸不保守。
| 图表编号 | XD0014418000 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2018.04.15 |
| 作者 | 李华、邱建武、林桂枝、邓梅、林伟霞、程映、宋元宗 |
| 绘制单位 | 暨南大学附属第一医院儿科、暨南大学附属第一医院儿科、暨南大学附属第一医院儿科、暨南大学附属第一医院儿科、暨南大学附属第一医院儿科、暨南大学附属第一医院儿科、暨南大学附属第一医院儿科 |
| 更多格式 | 高清、无水印(增值服务) |


{kind=link}