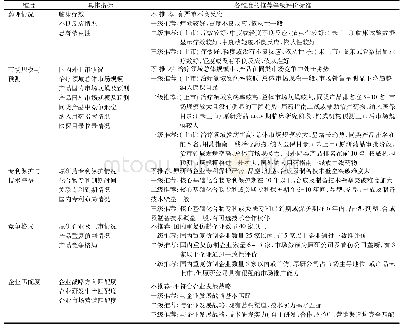
《表1 我国仿制药一致性评价相关政策法规》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
我国一致性评价政策可以划分成三个阶段[5](表1)。第一阶段是启动阶段:2012~2014年,仿制药一致性评价由2012年“十二五”规划提出,为分时期和分阶段评价奠定了基础,于2013年正式推广,但是由于缺乏确定的评价体系导致评价工作处于停滞期;第二阶段:2015~2016年,全面开启一致性评价工作,这一时期有两个重要的文件出台,2015年《关于改革药品医疗器械审评审批制度的意见》(国发(2015) 44号)文件对一致性评价工作产生推动作用,提出要加快评价工作进度,2016年《关于开展仿制药质量和疗效一致性评价的意见》(国办发(2016) 8号)文件明确评价的具体目标与时间,一致性评价才正式开始实施;第三阶段是2017年以后,为政策突破期,其中2017年《关于仿制药质量和疗效一致性评价工作有关事项的公告》(2017年第100号)文件对一致性评价相关事项进行了详细说明[6],更好地指导评价工作的开展,随后还发布了注射剂的征求意见稿,注射剂的一致性评价工作在2020年正式启动。从2012年至今,我国对一致性评价工作越来越重视,各项政策法规不断出台,一致性评价体系逐渐完善,越来越科学规范,从评价方法、参比制剂、临床试验等方面不断细化,为企业提供指导。评价方法从主要是体外溶出度试验变为以生物等效性(BE)试验为主,而对于满足BE豁免政策的品种可以不进行BE试验而是采用体外试验如体外溶出度等来证明生物等效性,并且出台了仿制药参比制剂目录,评价对象也逐步从289目录内品种扩展至上市时间在新注册分类实施前的仿制药[7]。
| 图表编号 | XD00199146700 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2020.10.28 |
| 作者 | 胡宇、宗欣、于淼、李勇 |
| 绘制单位 | 中国药科大学国际医药商学院、国家药品监督管理局信息中心、国家药品监督管理局信息中心、中国药科大学国际医药商学院 |
| 更多格式 | 高清、无水印(增值服务) |





{kind=link}