《表2 不同计算方法得到的最稳定的两体分子的电荷》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
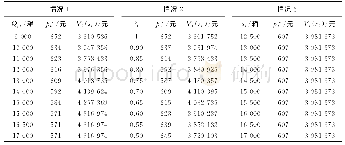
基于最稳定的两体分子,我们系统考察了不同计算方法对其带电荷的影响,结果列于表2。通过对比,我们发现计算方法能够影响堆积分子的带电荷性质。例如,在pyrene-OFN晶体的最稳定两体分子(Dimer A)中,pyrene分子在M06-2X/6-31G计算水平下为0.073 e,在M06-2X/6-31G(d,p)和M06-2X/6-31+G(d,p)计算水平下,其值分别为0.064 e和0.075 e。计算还表明,堆积分子在M06-2X/6-31+G(d,p)和M06-2X/6-311++G(d,p)计算水平下得到的电荷基本趋于不变。例如,在这两种计算水平下,pyrene-OFN最稳定两体中,pyrene分子的带电荷基本趋于0.075 e,而pyrene-TCNB最稳定两体中pyrene分子基本趋于0.092 e。因此,基于共晶中最稳定的两体结构,采用M06-2X/6-31+G(d,p)计算方法可以近似描述有机共晶中的电荷转移性质。
| 图表编号 | XD00153875600 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2020.08.01 |
| 作者 | 张德超、刘国魁、冷霞、周广丽、李云志、夏其英 |
| 绘制单位 | 临沂大学化学化工学院、临沂大学化学化工学院、临沂大学化学化工学院、临沂大学化学化工学院、临沂大学化学化工学院、临沂大学化学化工学院 |
| 更多格式 | 高清、无水印(增值服务) |





{kind=link}