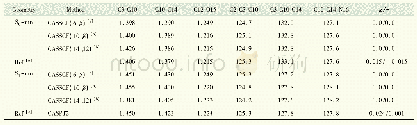
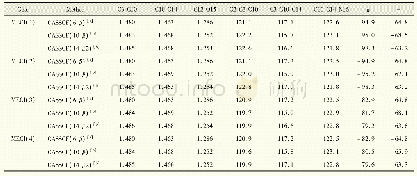
《表1 在不同的计算水平下优化得到的HBI-分子在不同电子态的重要结构参数(键长单位为10-1pm,键角和二面角的单位为(°))》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
本系列图表出处文件名:随高清版一同展现
《关于绿色荧光蛋白发色团激发态动力学行为的实验设计与实践》
[a]6-31G基组;[b]6-31G*基组;[c]参考文献13中的数据
实验中发现更高能级的电子激发态对发色团分子的退激发行为影响甚微,因此在计算过程中只考虑了基态(S0)和第一电子激发态(S1)[18-19]。所有的电子结构计算都采用Molpro软件中的CASSCF方法。CASSCF计算中有两个具有相同比重的电子态参与态平均。值得一提的是,CASSCF的计算方法依赖于活化空间的选择,因此选择合适的活化空间至关重要。一般情况下,选择的活化空间越大,计算结果越准确,但是计算耗时也越长。所以在进行静态电子结构计算时,优先选择大的活化空间和基组以保障计算的准确度。但是对于动态的动力学模拟过程,选择大的活化空间和基组会消耗大量的计算机时,因此还需要在保证计算精度的前提下,选择计算耗时更短的小活化空间。为此,对比了不同活化空间(14e,12o)、(10e,8o)以及(6e,5o)和不同基组下(6-31G*和6-31G)优化的稳定构型和锥形交叉点的结构参数,如表1、2所示。通过数据可见,(6e,5o)的活化空间搭配6-31G的基组优化得到的构型参数与大活化空间(14e,12o)和6-31G*基组得到的相差不大。另外,由于分子的激发态行为取决于该分子激发态和基态势能面的拓扑结构,为了进一步确认(6e,5o)/6-31G计算的准确性,利用线性插值法LIIC得到了Franck-Condon点与锥形交叉点间的势能曲线,如图4所示。二者的拓扑结构一致,说明用(6e,5o)/6-31G进行动力学模拟得到的结果是可信的。
| 图表编号 | XD00172316300 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2020.02.25 |
| 作者 | 赵莉、郑海霞、展凯云、刘冰 |
| 绘制单位 | 中国石油大学(华东)理学院、中国石油大学(华东)理学院、中国石油大学(华东)理学院、中国石油大学(华东)理学院 |
| 更多格式 | 高清、无水印(增值服务) |

![表1 优化的[An L]–和[An L]几何参数(键长单位为nm,键角单位为(°))](http://bookimg.mtoou.info/tubiao/gif/HXXB202010010_01100.gif)


{kind=link}