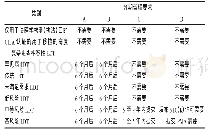
《表1 FDA对LDT分级与分期管理的要求》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
注:A为上市前通知/注册和列名(FD&CA510;21CFR807),B为医疗器械不良事件报告[FD&CA519(a);21CRF803E],C为上市前审查[FD&CA510(k),515;21CFR807E,814],D为质量系统法规[FD&CA520(f);21CFR820]。时间以FDA指南定稿颁布日期为准,表2同。
在FDA的LDT监管框架指南(草案)中,将LDT作为IVD的一个子集进行监管,其核心是分级与分期,即将LDT基于风险进行分级,分阶段逐步推行管理措施,并对特定类型LDT的特定监管要求执行执法自由裁量权(见表1和表2)。首先,FDA认为与LDT有关的健康风险和所有IVD一样,因每种诊断试剂的不同而异,所以监管活动应相应地基于风险执行;其次,分阶段的执行能缓解立即执行所有适用要求的非预期后果,如潜在的临床检测试剂供应短缺。FDA将根据风险分类,优先完成高风险LDT的上市前审查,再逐步推进中、低风险LDT的监管。在进行LDT对患者或消费者构成的风险评估时,FDA考虑的因素包括:1) LDT是否会用于高风险疾病患者群;2) LDT用于疾病筛查还是诊断;3) 基于LDT结果进行的临床决策性质;4) 医师或病理学家是否会有除LDT结果以外的关于患者的信息来帮助进行临床决策;5) 患者可获得备选的诊断和治疗方案;6) 错误结果的潜在影响;7) 与LDT相关的不良事件的数量和类型等。
| 图表编号 | XD0081568000 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.06.20 |
| 作者 | 施一然、梁毅 |
| 绘制单位 | 中国药科大学国际医药商学院、中国药科大学国际医药商学院 |
| 更多格式 | 高清、无水印(增值服务) |





{kind=link}