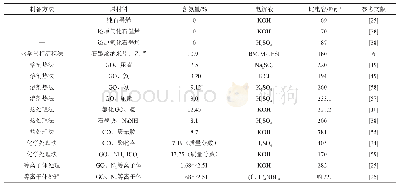
《表1 N、P、S掺杂石墨烯与铀酰结合体系的吸附能》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
本系列图表出处文件名:随高清版一同展现
《氮、磷、硫共掺杂石墨烯材料对环境中铀酰去除机理的理论模拟研究》
对于G(NP),与N原子相比,P原子周围呈较强的负电势,是亲电基团的主要进攻位点.U原子带两个正电荷,因此当铀酰的吸附位点为U原子时,N&P共掺杂石墨烯对铀酰吸附起主要作用的原子为P原子,如图4(a1~c1)所示,无论N、P的相对位置为对位、间位、邻位,U原子与P原子之间的键长都在较短的范围内,分别为3.161、3.007、3.047?.它们的吸附能分别为1.49、15.59、4.69 kcal/mol(表1).当铀酰的作用位点为Oax时,如图4(a2~c2)所示,其与P原子之间的距离分别为3.734?([G (NP-para)···O=U=O]) 、3.692?([G (NP-meta)···O=U=O]) 、3.669?([G (NP-ortho)···O=U=O]) ,略长于相应的P–U键长.其中的N–Oax键长分别为3.301、4.542、3.279?.且当铀酰的作用位点为Oa x时,计算所得的吸附能为9.7 7、5.6 3、3.67 kcal/mol,总体上相比[G(NP)···UO2]较低.说明当铀酰的作用位点为Oax时,与G(NP)的相互作用较弱.另一方面,在[UO2(H2O)5]2+中,U原子与其轴向氧之间的键长(U=Oax)为1.732/1.733?,而在[G(NP-para)···UO2]、[G(NP-meta)···UO2]、[G(NP-ortho)···UO2]中,由于铀酰与N&P共掺杂石墨烯的相互作用,U=Oax之间的键长被不同程度地拉长了,分别变为1.740/1.739、1.748/1.747、1.750/1.732?.但当铀酰与N&P共掺杂石墨烯的作用位点为Oax时,U=Oax的变化较小,进一步说明当铀酰的作用位点为U原子时,与N&P共掺杂石墨烯的相互作用更强.
| 图表编号 | XD0034636000 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.01.20 |
| 作者 | 柳杨、赵超锋、张安睿、邢金璐、卫冬丽、艾玥洁 |
| 绘制单位 | 华北电力大学环境科学与工程学院、华北电力大学环境科学与工程学院、华北电力大学环境科学与工程学院、华北电力大学环境科学与工程学院、华北电力大学环境科学与工程学院、华北电力大学环境科学与工程学院 |
| 更多格式 | 高清、无水印(增值服务) |





{kind=link}