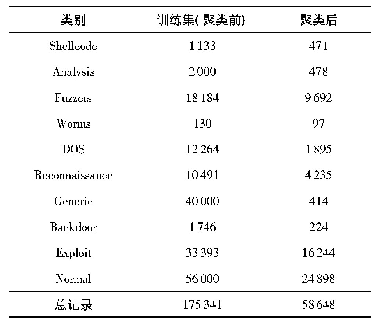
《表1:数据集样本数量:石墨烯与色氨酸相互作用模式的分子模拟研究》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
本次对接我们采用的是刚性对接的对接方法,即石墨烯与氨基酸的对接只在空间位置和姿态上发生了改变,结构上并无变化。关于对接过程,我们共进行了300次的对接计算,平均分为A、B、C三组,每组100次。在分子对接理论中,能量越低,分子结合越稳定,因此我们采用筛选能量的方法进行最优结合构象的选择。在这三组中,首先我们对每一组进行单独分析,根据Auto Dock的程序设定,综合能量越低的优势构象会在结果播放窗口中越靠前,因此我们选择了每组数据的第一组构象作为本组最优并进行能量数据分析,见下表1。
| 图表编号 | XD00205872900 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2020.11.01 |
| 作者 | 童明琼、刘鹏、孙婉、范娜、王晓玥 |
| 绘制单位 | 德州学院医药与护理学院、德州学院、德州学院医药与护理学院、德州学院医药与护理学院、德州学院医药与护理学院 |
| 更多格式 | 高清、无水印(增值服务) |



{kind=link}