《表1 催化剂结构的形成能和具体计算参数》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
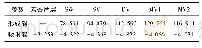
大量的研究已经表明,单原子铁掺杂含氮碳材料的催化活性中心是Fe N4位点,即铁单原子与四个吡啶氮配位,如图1所示[16].因此,我们研究的Fe-NC催化剂采用与文献中相同的Fe N4模型,在此基础上研究S原子掺杂的可能构型.根据Li等[10]的研究,S原子和N原子的比例为n(S)∶n(N)=1∶1时,Fe NS催化剂催化氧还原反应的活性是最高的,为了研究S掺杂比例的影响,我们考虑了1~4个S原子掺杂的构型,如图1所示,一个S原子掺杂有两种可能的构型,分别为S原子与N原子成键(Fe NS1a)和S原子不与N原子成键(Fe NS1b).形成能的数值小于零,表明该结构是热力学稳定的,形成能的数值大于零,表明该结构是不稳定的.根据形成能计算(表1),Fe NS1a的形成能为-0.06 e V,Fe NS1b的形成能为0.50 e V,结构Fe NS1a的稳定性优于结构Fe NS1b,因此2~4个S原子掺杂的Fe NS只考虑S原子与N原子直接成键结构.在Fe NS2结构中,引入两个S原子的形成能为-0.27 e V,表明其热力学稳定性优于Fe NS1a结构,进一步提高S原子掺杂的量,可以进一步提高催化剂结构的稳定性,在Fe N4位点引入四个S原子掺杂时,结构的形成能为-1.74 e V.形成能的计算结果表明,在Fe N4位点周围引入S原子掺杂可以提高催化剂结构的稳定性.通过计算Fe—N键长发现,硫原子掺杂对Fe—N键长有较大影响.Fe N4结构中Fe—N键的键长为0.188 nm,而Fe NS1a和Fe NS1b中Fe—N的键长分别为0.183 nm和0.189 nm,Fe NS2结构中Fe—N的键长为0.183 nm.Fe NS1a和Fe NS2构型中Fe—N键长明显缩短,此时所有的原子仍然在同一平面内.在Fe NS3结构中,Fe—N键长为0.187 nm,Fe NS4结构中,Fe—N键长为0.201 nm,此时,由于S原子的原子半径大于氮原子,所以过多S原子的掺杂导致空缺位的出现,此时,其中一个S原子与N原子断键.
| 图表编号 | XD00195241100 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2020.09.15 |
| 作者 | 鲁效庆、曹守福、魏晓飞、李邵仁、魏淑贤 |
| 绘制单位 | 中国石油大学材料科学与工程学院、中国石油大学材料科学与工程学院、中国石油大学材料科学与工程学院、中国石油大学材料科学与工程学院、中国石油大学材料科学与工程学院 |
| 更多格式 | 高清、无水印(增值服务) |





{kind=link}