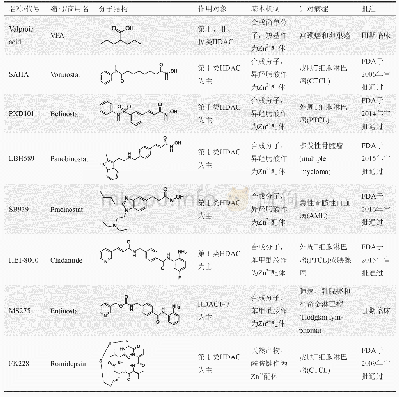
《表2 以苯甲酰胺为ZBG的HDAC抑制剂》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
本系列图表出处文件名:随高清版一同展现
《组蛋白去乙酰化酶抑制剂中锌离子结合基团的研究进展》
苯甲酰胺类HDAC抑制剂与异羟肟酸类相比,对第Ⅰ类HDAC具有更高的选择性。药物-受体的作用模型研究表明,苯甲酰胺中的羰基和氨基可以与HDAC中的锌离子螯合,在氨基对位引入氟原子,可以增强对HDAC3的选择性,而引入空间体积较大的基团如噻吩,可以增强对HDAC1和2的选择性。西达本胺(tucidinostat/chidamide,6)已于2014年被原国家食品药品监督管理总局(CFDA)批准用于治疗HR阳性/HER2阴性乳腺癌和外周T细胞淋巴瘤[21],恩替诺特(entinostat/MS-275,7)、mocetinostat(MGCD0103,8)和domatinostat(4SC-202,9)目前正处于Ⅱ期临床研究[22],而tacedinaline(CI-9943,10)的Ⅱ期临床试验已终止[23],见表2。相比较于异羟肟酸类,苯甲酰胺类HDAC抑制剂具有较高的Ⅰ类HDAC选择性,而HDAC抑制剂的不良反应会随着选择性的提高而降低,因此,苯甲酰胺类HDAC抑制剂具有较低的毒副作用。虽然高选择性意味着临床试验中的高安全性,但苯甲酰胺结构中暴露的氨基可能会潜在地诱导体内毒性的产生[24]。此外,肿瘤细胞更有可能对单一靶点的高选择性抑制剂产生耐药性,因此可能会影响苯甲酰胺类HDAC抑制剂在长期治疗中的效果。针对这一可能出现的问题,现有2种策略可以用来解决该问题:一种是HDAC抑制剂和其他靶点抗癌药物联合用药;另外一种开发多靶点的HDAC混合抑制剂[25]。
| 图表编号 | XD00181780000 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2020.05.30 |
| 作者 | 朱玉垚、王瑜、马俊杰 |
| 绘制单位 | 华侨大学医学院、华侨大学医学院、华侨大学医学院 |
| 更多格式 | 高清、无水印(增值服务) |




{kind=link}