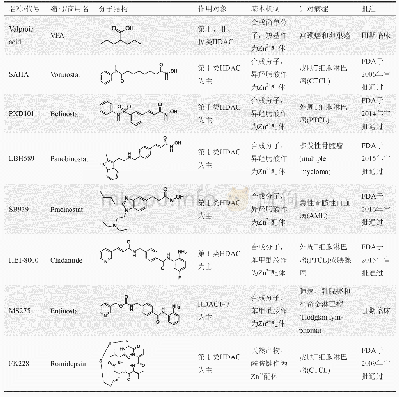
《表3 针对第Ⅰ、Ⅱ和Ⅳ类HDAC的抑制剂代表》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
由于第Ⅰ、Ⅱ和Ⅳ类HDAC的活性位点都具有Zn2+,故而对其的主要抑制机制便是有机官能团对Zn2+的配位。对此,最为简单的抑制剂丙戊酸(valproic acid,VPA)直接利用其羧基即可实现抑制,且处于宫颈癌和卵巢癌的Ⅲ期临床试验阶段。其他一些结构复杂的抑制剂则可大致分为两类:第一类基于异羟肟酸(hydroxamic acid)结构基元,包含SAHA、PXD101和LBH589,这三者分别由美国食品与药品监管局(FDA)于2006、2014和2015年审批成药;第二类包含苯甲酰胺(benzamide)结构基元,代表物质为FDA于2015年审批通过的HBI-8000和尚处于治疗肺癌和乳腺癌Ⅱ期临床试验阶段的MS275。此外,还有一些天然产物也同样可以抑制HDAC,如FK228于2009年通过FDA审批作为皮肤T细胞淋巴瘤药物(cutaneous T-cell lymphoma)。这些抑制剂的具体信息如表3[45]。
| 图表编号 | XD00207037900 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2020.12.01 |
| 作者 | 王志鹏、张璇、蒋振雄、车子良、马新雨 |
| 绘制单位 | Division of Genetics, Department of Medicine, Brigham and Women's Hospital, Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School、Department of Chemistry, Texas A&M University、清华大学化学系、清华大学生命学院、Department of Plant and Patho |
| 更多格式 | 高清、无水印(增值服务) |
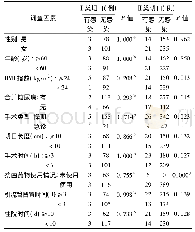
![表3 Ⅰ、Ⅱ类切口手术预防性抗菌药物使用率和联合用药率[n(%)]](http://bookimg.mtoou.info/tubiao/gif/ZYYG202017063_01700.gif)




{kind=link}