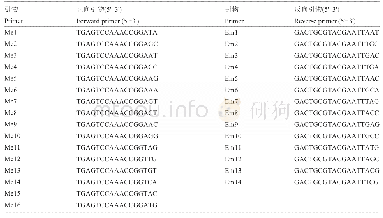
《表1 用于分子生物学鉴定的引物及标签码序列》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
对于分离获得的纯菌株使用改良的十六烷基三甲基溴化铵(CTAB)法提取基因组DNA(Zhang et al.2017)。用于高通量测序的底泥样品宏DNA提取方法参照FastDNA Spin Kit试剂盒(MP Biomedicals,美国)说明书。1.4.2真菌ITS2区PCR扩增:由各分离培养方法获得的纯菌落DNA和未经培养的沉积物宏DNA均使用ITS2区进行PCR扩增分析。由于原位培养方法会获得大量纯菌落,为提高鉴定效率,降低测序及时间成本,本研究设计了24对标签码(表1),分别连接在正反向引物的5’端,共计576种组合,用于区分各个纯菌落。基因组DNA经无菌水稀释至1ng/μL作为模板,使用经标签码修饰的ITS7/ITS4(Karlsson et al.2014)引物进行扩增。在25μL反应体积中包括10×PCR缓冲液2.5μL,MgCl2 2mmol/L,dNTPs 50μmol/L,正向和反向引物各0.1μmol/L,DNA聚合酶0.5U,DNA模板10ng。反应条件:94℃10min;95℃30s,53℃30s,72℃1min,共进行35个循环;72℃10min。PCR产物经1%琼脂糖凝胶电泳检测合格后,送至微分(上海)基因科技有限公司测序。将所有培养得到菌株的代表序列在NCBI的GenBank数据库中进行比对,以97%相似度为阈值将各菌株注释到属。若注释到多个属,以前20条结果中某属出现的顺序、数量、序列覆盖率、序列可信度结合菌株自身形态特征确定。1.4.3生物信息学分析:利用USEARCH 10软件测序对获得的真菌ITS2原始数据进行处理和分析(Edgar 2013)。根据每个样本所对应的barcode序列拆分获得的每个样本的有效序列。使用USEARCH10中的fastq_filter命令对所有FASTQ格式的双端序列进行质量筛查以过滤掉低质量的序列,保证碱基平均测序准确率≥99%。使用fastq_mergepairs命令进行序列双端合并将双端测序数据合并成完整序列。通过USEARCH10中的UCHIME命令比对到UNITE UCHIME数据库(https://unite.ut.ee/repository.php)(Nilsson et al.2015)进行嵌合体的查找和剔除。通过使用UPARSE(≥97%)聚类算法对去掉嵌合体后的所有序列进行聚类和OTUs划分,并选取OTUs中丰度最高的序列作为其代表性序列。最后用RDP分类算法(Rognes et al.2016)对所有代表性序列进行物种注释并将不能注释到任何已知分类单元的序列定义为未鉴定OTU(unidentified)。对OTU丰度矩阵中的所有样本用vegan包进行稀释曲线的计算,以评估是否所有的样品的测序深度都达到了平台期(Oksanen et al.2015)。使用USEARCH对OTUs表中的所有样品按照最少测序量的样品数据量进行统一序列量抽平处理,从而减少由于不同样品测序深度造成的多样性差异。分别计算每个样本香农指数(Shannon)以及均匀度(Pielou)指数指示物种多样性和均匀度。香农指数是用来估算样本中微生物多样性指数之一,值越大说明群落多样性越高。均匀度指数是反映不同物种分布的均匀度,值越大说明均匀度越高。所有分析均在R v.3.2.5(Core Team2016)中进行。
| 图表编号 | XD00163525500 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2020.04.22 |
| 作者 | 周士越、周欣、张志锋、梁俊敏、蔡磊 |
| 绘制单位 | 中国科学院微生物研究所真菌学国家重点实验室、中国科学院大学生命科学院、中国科学院微生物研究所真菌学国家重点实验室、中国科学院大学生命科学院、中国科学院微生物研究所真菌学国家重点实验室、中国科学院大学生命科学院、中国科学院微生物研究所真菌学国家重点实验室、中国科学院微生物研究所真菌学国家重点实验室、中国科学院大学生命科学院 |
| 更多格式 | 高清、无水印(增值服务) |





{kind=link}