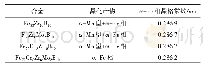
《表1 Bi2Te3-xSex(x=0,1,2,3)四种材料实验的和优化后的晶格常数》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
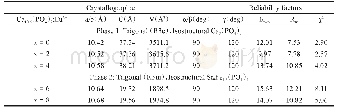
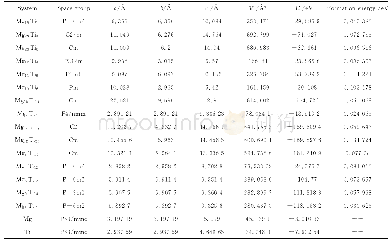
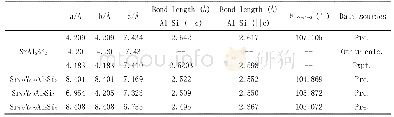
Se与Te是同族元素,计算时通过Se原子替换Te原子实现Se掺杂Bi2Te3。由于Se原子的电负性比Te原子的强,实验中进行掺杂时Se原子通常先占据Te(2)位,再占据Te(1)位。因此计算时也是先替换Te(2),再替换Te(1),依次得到Bi2Te2Se、Bi2Te1Se2、Bi2Se3三种结构,它们都是Bi2Te3的同系化合物,晶格结构与Bi2Te3类似。由于用第一性原理计算方法计算的是晶体在理想状态下的性质,所以在进行理论计算时不能直接采用实验上得到的晶格常数值,而是要先进行结构上的优化。通过结构优化可以找出理论上使晶体具有最低能量的晶格常数。表1列出了各结构实验的和优化后的晶格常数。可以看到随着Se掺杂浓度的增大,Bi2Te3-xSex的晶格常数a和c逐渐减小,相应的它们的原胞体积也在减小。这主要因为Se的原子半径比Te小,导致Se掺杂后Bi2Te3-xSex的原胞体积收缩。与实验晶格常数值[9]比较,可以看到我们计算优化出来的晶格常数较实验值稍微偏大,这主要是因为密度泛函计算中PBE交换关联能通常会高估材料的晶格常数。但我们也可以看到,理论值与实验值误差在3%以内,并且计算结果给出的Bi2Te3-xSex的晶格常数随Se掺杂浓度升高而减小的变化趋势与实验结果是一致的。
| 图表编号 | XD0092442600 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.06.30 |
| 作者 | 邹彦娟、张小龙 |
| 绘制单位 | 萍乡学院机械电子工程学院、萍乡学院机械电子工程学院 |
| 更多格式 | 高清、无水印(增值服务) |


{kind=link}