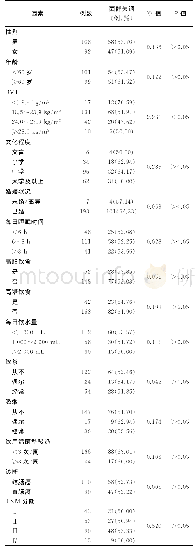
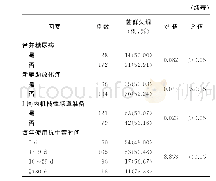
《表1 肠道微生物群研究相关的工具/网络服务器》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
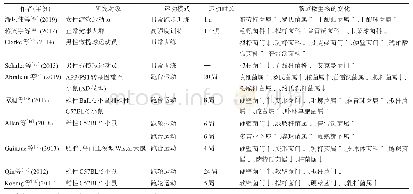
为了解决分析宏基因组数据中遇到的困难,目前已经开发出Web服务器和R软件包,并且可以在公共领域中进行使用。许多独立的工具能够分析16S rRNA标记基因测序数据和WGS数据,如定量洞察微生物生态学(quantitative insights into microbial ecology,QIIM E),使用16S rRNAs数据研究微生物多样性。这些独立工具可为用户提供系统发育分析的分类指示,以及解析和过滤从Illumina或其他平台生成的原始数据。但是QIIM E的安装需要一些Linux和Windows系统的专业知识,并且在操作分类单元(operational taxonomic units,OTU)选择步骤中缺乏并行处理功能[27]。M othur是一个具有多种功能的软件包,包括OTU的识别和不同样本之间的α(在特定样本中)和β(不同样本之间)差异的描述[28]。RAMMCAP是一个基于图形用户界面(graphical user interface,GUI)的工具,它可对宏基因组序列进行聚类和分析,与其他工具和软件相比,可以在很短的时间内处理大量序列。RAMMCAP还包括蛋白质家族注释工具和基于统计分析的新型GUI的宏基因组比较方法[29]。对于基于WGS的测序数据分析(主要用于分类学分类),有几种方法可用,它们集成了基本局部比对搜索工具(basic local alignment search tool,BLAST)用于物种鉴定。M EtaGenome ANalyzer(M EGAN)使用BLAST搜索参考序列数据库,如来自NCBI NR数据库的非靶向序列数据库,并在GUI中提供结果;它允许分析大型数据集而无需进一步组装或靶向特定的16S rRNA标记基因;它还可以基于统计分析比较不同的数据集,并提供输出图形[30]。此外,一些软件包也可以同时进行组装和注释,例如MOCAT,它将宏基因组短读段组装成重叠群,同时进行质量控制,并从重叠群进行基因预测[31]。对于宏基因组读数的功能分析,可以使用来自组装的重叠群的预测基因或具有一定浏览长度的原始序列读数。某些途径,如Smash Community[32]、微生物群项目统一代谢分析网络(HUMANN)[33]以及宏基因组的功能注释和分类学分析(functional and taxonomic analysis of metagenomes,FANTOM)[34],它们是用于宏基因组数据分析的易于使用的GUI,也可用于自动化装配和注释过程(表1)。
| 图表编号 | XD0086142800 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.09.01 |
| 作者 | 赵磊、孙会、马燕芬 |
| 绘制单位 | 吉林农业大学动物科学与技术学院吉林省动物营养与饲料科学重点实验室、吉林农业大学动物科学与技术学院吉林省动物营养与饲料科学重点实验室、内蒙古农牧业科学院动物营养研究所 |
| 更多格式 | 高清、无水印(增值服务) |



{kind=link}