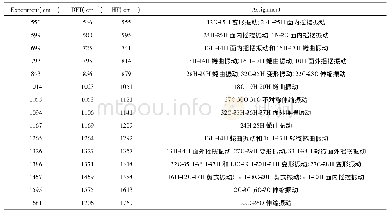
《表2 M06-2X/6-311++G(d,p)计算水平下得到的化合物1o,1p和1q中性和质子化形式的能量以及对H+的亲和能,其中以H+作为能量零点》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
本系列图表出处文件名:随高清版一同展现
《"“受阻路易斯酸碱对”催化的2,3-二取代2H-1,4-苯并噁嗪氢化反应机理研究"》
此外,我们也直接计算了化合物1o,1p和1q对H+的亲和能力以评价N4位点碱性的强弱,我们以H+为能量零点,分别计算了化合物1o,1p和1q的中性形式以及N4位点被质子化后阳离子形式的自由能,得到了质子亲和能(PA).如表2所示,化合物1o,1p和1q对于H+的亲和能分别为254.6,255.0和255.1 kcal/mol,几乎相同,表明这3种化合物中N4位的碱性基本相同,这和我们通过NBO计算及Mulliken电荷分析得到的N4位点电荷分布结果一致.因此可以推测在FLPs催化的2,3-二取代2H-1,4-苯并噁嗪氢化反应中3号位取代基的位阻效应对反应起主要作用,而电子效应几乎可以忽略.
| 图表编号 | XD003166100 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.03.15 |
| 作者 | 魏思敏、王英辉、赵红梅 |
| 绘制单位 | 陕西中医药大学陕西省中药资源产业化协同创新中心、中国科学院化学研究所北京分子科学国家实验室、中国科学院化学研究所北京分子科学国家实验室 |
| 更多格式 | 高清、无水印(增值服务) |
![表2 胃黏膜肠化情况对患者血清PG和G-17水平的影响[M(P25,P75)]](http://bookimg.mtoou.info/tubiao/gif/ZWJZ202020018_02600.gif)
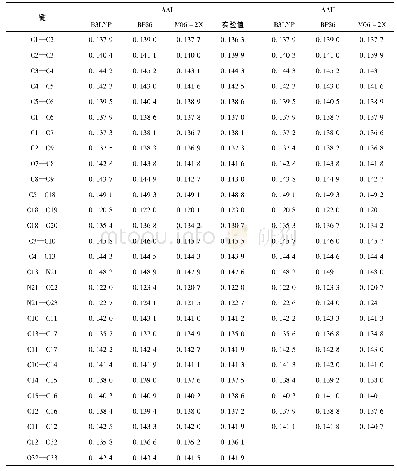
![表6 在M06-2X/6-311G(d,p)水平下计算得到的[HEtHex][Acylala]的氢键BCP的电子密度性质](http://bookimg.mtoou.info/tubiao/gif/HGSZ2020S1003_03200.gif)
![表5 在M06-2X/6-311G(d,p)水平下优化得到[HHex][TFS]-n H2O分子间的相互作用BCPs的电子密度(kcalmol-1)](http://bookimg.mtoou.info/tubiao/gif/HGSZ2019S2015_03600.gif)
{kind=link}