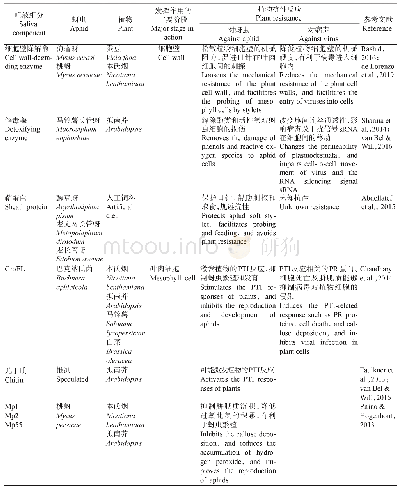
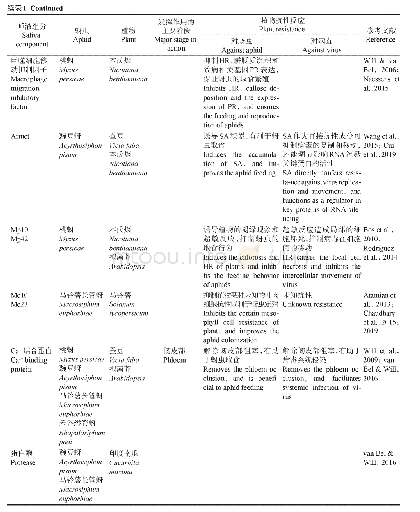
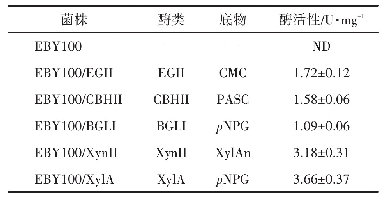
《表1 病毒组分与炎性小体的组装》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
病毒入侵机体后,会与相应的模式识别受体结合,进而诱导炎性小体的激活(表1)。研究发现,呼吸道合胞病毒(RSV)编码的疏水蛋白SH在RSV感染诱导NLRP3炎性小体激活中发挥重要作用[38]。RSV感染后,RSV病毒的SH蛋白聚集在高尔基体的脂筏结构中,形成离子通道,促使NLRP3从细胞质向高尔基体的移位,诱导了炎性小体的活化。而用抑制病毒离子通道和脂筏解离的药物对RSV感染的细胞进行药物治疗后,则阻断了炎性小体的激活,表明SH蛋白在高尔基体内形成的脂筏结构在炎性小体激活中起重要的作用[38]。有些病毒还会利用自身编码的非结构蛋白拮抗炎性小体的形成,以利于自身的复制。如人巨细胞病毒(HCMV)编码的pUL83蛋白通过自身的PAD结构域与IFI16蛋白的PYD结构域结合,抑制IFI16多聚化,从而抑制IFI16炎性小体的活化[39]。麻疹病毒(MV)V蛋白直接与NLRP3蛋白相互作用,抑制NLRP3炎性小体介导的IL-1β活化[40]。肠道病毒71型(EV-71)的2A和3C蛋白酶分别在Q225-G226或G493-L494上切割NLRP3蛋白,以抑制NLRP3炎性小体的激活[41]。此外,EV-71的3C蛋白酶还被证明能在Q193-G194上切割GSDMD,在氨基端产生更短的GSDMD片段,从而抑制细胞焦亡,促进EV-71的复制[42]。甲型流感病毒(IAV)的非结构蛋白N S 1在病毒感染巨噬细胞的过程中,通过抑制caspase-1的激活来限制成熟形式的IL-1β和IL-18的产生[30]。缺乏完整NS1基因的突变病毒感染巨噬细胞后,IL-1β和IL-18的表达水平明显上升。同时,氨基端包含RNA结合域,羧基端缺失效应域的NS1突变体病毒,能拮抗IL-1β和IL-18的产生,说明IAV编码的NS1蛋白通过其氨基末端和羧基末端结构域的功能控制感染的巨噬细胞中促炎性细胞因子的产生[43]。重症急性呼吸综合征冠状病毒(SARS-CoV)的辅助蛋白ORF3a通过与肿瘤坏死因子受体相关因子3(TRAF3)相互作用,促进ASC泛素化,导致caspase-1活化和IL-1β成熟,从而激活NLRP3炎性小体[44]。
| 图表编号 | XD00225421000 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2020.08.01 |
| 作者 | 武雪宁、李素、曹宏伟、仇华吉 |
| 绘制单位 | 黑龙江八一农垦大学生命科学技术学院、中国农业科学院哈尔滨兽医研究所兽医生物技术国家重点实验室、中国农业科学院哈尔滨兽医研究所兽医生物技术国家重点实验室、黑龙江八一农垦大学生命科学技术学院、中国农业科学院哈尔滨兽医研究所兽医生物技术国家重点实验室 |
| 更多格式 | 高清、无水印(增值服务) |



{kind=link}