《表1 8种化学结构键阿莫西林5-S相关电荷计算》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
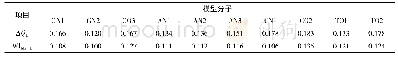
本论文首先使用Chem Draw8.0模块和Chem Draw 3D软件模块,构建了8个阿莫西林及其衍生物分子的模型,运行软件并使其结构和能量最低化;在Gauss View 5.0.9软件模块中,使用B3LYP泛函方法在6-31+G(d,p)基组水平下优化其分子结构,我们的计算关键词为pop=(nbo,chelpg,hirshfeld)的约束条件下,分别计算出8个阿莫西林衍生物的DFT和DFRT量化参数。记录5-S的Mulligen、ESP、NBO和HIR的电荷值;运用ACD Labs6.0软件,得到8种阿莫西林衍生物相应水溶液中的溶解度(log S)预报值;然后分别将8种阿莫西林及其衍生物的Mulligen电荷、ESP电荷、NBO电荷和HIR的电荷值拟合,用Origin 8.5作图,得到阿莫西林及其衍生物溶解度(log S)相应的线性回归方程,比较R值(相关系数)大小,来确定R值最大的就是预测方程。由表1可知,阿莫西林椅式结构上H的平伏键变成直立键时,log S保持不变。这说明化学结构键的变化对于阿莫西林的溶解度没有影响;阿莫西林椅式结构上H的平伏键变成直立键时,5-S的Mulligen、NBO和HIR的电荷值存在较小的变化,而5-S的ESP电荷值变化明显(y=13.51x-0.72,R2=0.912)。
| 图表编号 | XD00208697700 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2020.07.28 |
| 作者 | 洪文燕、施琪浩、张英杰、蒋鑫、陈诗雨、章少阳、陈茹箐、秦利明、钟爱国 |
| 绘制单位 | 台州学院医药化工与材料工程学院、台州学院医药化工与材料工程学院、台州学院医药化工与材料工程学院、台州学院医药化工与材料工程学院、台州学院医药化工与材料工程学院、台州学院医药化工与材料工程学院、台州学院医药化工与材料工程学院、台州学院医药化工与材料工程学院、台州学院医药化工与材料工程学院 |
| 更多格式 | 高清、无水印(增值服务) |


![表3[HAlkyl][TFSA]S1~S5结构中的NPA计算的电荷分布](http://bookimg.mtoou.info/tubiao/gif/SHZN202005004_03500.gif)
![表3[HAlkyl][TFSA]S1~S5结构中的NPA计算的电荷分布](http://bookimg.mtoou.info/tubiao/gif/SHZN202005004_03501.gif)

{kind=link}