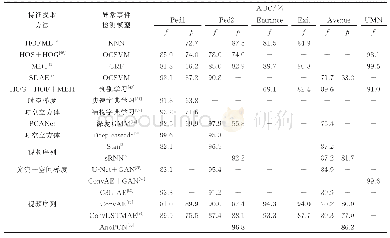
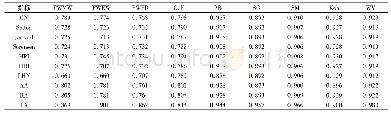
《表1 不同方法的AUC值》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
为了评估所提方法的性能,使用了来自DNA元素百科全书ENCODE项目的125个TF结合位点ChIP-seq实验,包括A549,MCF-7,H1-HESC和HUVEC。对于每种细胞类型,从峰文件中的每个记录中选择居中的101 bps作为阳性样品。为了满足模型测试要求,通过匹配正样本的大小,GC含量和重复分数,生成了相等数量的负样本。每个数据集随机分为3组:训练、验证和测试集。为了训练k-mer嵌入模型,通过将k设置为5,将跨度s设置为2来生成k-mer语料库。在Gensim包中使用Word2vec模型的Python实现来获取k-mer嵌入向量。Word2vec中的所有参数均保留其默认值。将所提方法与3种其他方法进行比较:有gkmSVM,DeepBind和CNN_ZH的超参数保持不变。如表1所示。
| 图表编号 | XD00150095400 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2020.07.18 |
| 作者 | 石文 |
| 绘制单位 | 陕西学前师范学院数学与统计学院系 |
| 更多格式 | 高清、无水印(增值服务) |

{kind=link}