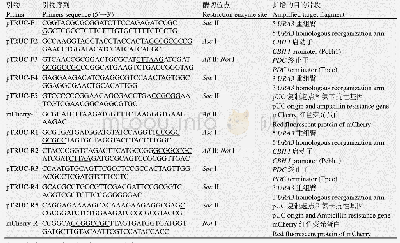
《表2 本研究所用引物:一个CRISPR/Cas9-VQR基因编辑系统的构建》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
本系列图表出处文件名:随高清版一同展现
《一个CRISPR/Cas9-VQR基因编辑系统的构建》
利用Cas9/VQR-gRNA靶点效率检测试剂盒(北京唯尚立德生物科技有限公司)检测4个gRNA对靶位点的切割效率。首先,参考试剂盒分别合成gRNA1、gRNA2、gRNA3和gRNA4引物(表2);然后以标准gRNA模板为模板,分别通过PCR扩增gRNA1、gRNA2、gRNA3和gRNA4及试剂盒提供的标准g RNA(S1和S2),回收纯化PCR产物作为DNA模板分别进行体外转录,回收并定量转录g RNA;其次,通过搭桥PCR的方式,将gRNA靶位点(包括PAM序列)构建到PCR产物中,获得用于酶切的DNA;最后,参照试剂盒提供的Cas9/VQR酶切体系,加入2μL 10×Cas9 buffer、1μL Cas9/VQR酶、50 ng体外转录的gRNA、50 ng酶切的DNA、补DEPC H2O至20μL,充分混合后,37℃反应30 min,然后加入3μL DNA Loading buffer,混合后65℃5min,2%琼脂糖凝胶电泳检测分析酶切结果。此外,每次酶切实验的同时做标准靶位点的gRNA酶切实验,以标准靶位点酶切实验结果作为阳性对照比较活性。通过与2个阳性参照标准S1和S2估算gRNA的活性。根据SSA活性,标准S1=3,标准S2=10,通过酶切条带的灰度与标准S1和标准S2对比,换算出gRNA1(g1)、gRNA2(g2)、gRNA3(g3)、gRNA4(g4)的活性(若检测gRNA酶切条带的灰度小于标准S1的灰度,则活性估值小于3;若检测gRNA酶切条带的灰度大于标准S1的灰度,且小于标准S2的灰度则活性估值大于3小于10;若检测gRNA酶切条带的灰度大于标准S2的灰度,则活性估值大于10)。
| 图表编号 | XD0078522200 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.06.12 |
| 作者 | 陈凯、孙国梁、宋高原、李爱丽、谢传晓、毛龙、耿帅锋 |
| 绘制单位 | 中国农业科学院作物科学研究所、中国农业科学院作物科学研究所、中国农业科学院作物科学研究所、中国农业科学院作物科学研究所、中国农业科学院作物科学研究所、中国农业科学院作物科学研究所、中国农业科学院作物科学研究所 |
| 更多格式 | 高清、无水印(增值服务) |





{kind=link}