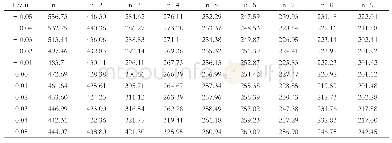
《表1 (TiO2) n (n=1~7) 团簇分子激发能 (E/eV) , 激发波长 (λ/nm) 和振子强度f》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
本系列图表出处文件名:随高清版一同展现
《(TiO_2)_n(n=1~7)纳米团簇激发态性质的基准计算》
从表1来看,与B3lyp计算出来的激发能相比较,Cam-b3lyp计算出来的激发能数值相对偏高,而PBEPBE泛函计算出来的激发能数值相对偏低。这是因为Cam-b3lyp是一种杂化泛函,该泛函考虑原子之间的长程相互作用,由于远程极高的HF成份,计算出的激发能也相对偏高。而PBEPBE泛函是典型的交换关联泛函,所含HF成份较低,因此计算出的激发能也偏低。与Enrico Berardo[8-9]利用精度更高的方法CCSD的计算结果比较,我们发现对于尺寸较小的团簇(n=1,2),B3lyp杂化泛函计算出来的激发能更准确。随着团簇尺寸增大,系统的电子态与原子核的振动态之间的耦合作用增强,因此考虑长程相互作用力的Cam-b3lyp泛函比B3lyp泛函能更好的描述(TiO2)n(n=1~7)团簇的激发态性质。因此我们建议,在计算尺寸较大的过渡金属团簇时,利用B3lyp进行结构优化,能够得到合理的基态结构参数;而对于激发态相关性质的计算,推荐使用含有HF成份较高的一些范围分离泛函,如Cam-b3lyp,能对其激发态的性质做一个合理的预测。
| 图表编号 | XD0072181800 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.06.01 |
| 作者 | 吴琪、霍炜 |
| 绘制单位 | 西藏大学理学院、西藏大学理学院 |
| 更多格式 | 高清、无水印(增值服务) |




{kind=link}