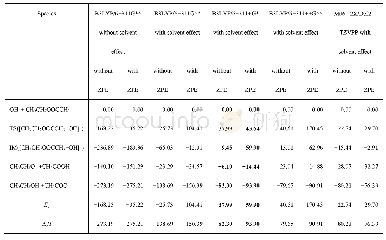
《表1 理论计算反应焓、中间体以及过渡态能量 (kJmol-1)》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
本系列图表出处文件名:随高清版一同展现
《将密度泛函理论计算纳入高等化学教学初探——对乙酸乙酯碱性水解机理的理论研究》
选择计算方法和基组的依据是:所得结构合理、结果符合已有实验或文献。本反应考查了几种计算方法和基组并将结果列于表1。由表中数据可以看出,在B3LYP[10]/6-311G**水平,不考虑溶剂效应计算得到的过渡态和活性中间体的能量都远远低于反应物,而考虑溶剂效应后,相应的能量仍低于反应物能量,这不符合图1所示的反应机理以及实验推测结果。究其原因,反应的过渡态和活性中间体都是负一价的阴离子,分别具有重要的弱相互作用和价电子分布,需要使用具有较小指数的高斯函数(也即弥散基)进行描述。6-311G**基组中G表示高斯基函数,6表示6个高斯基函数描述内层轨道,311表示3、1和1的三层劈裂的高斯基函数描述价层轨道,**表示C、O和H原子的极化高斯基函数,它们能描述原子轨道形成分子/离子的变化以及轨道空间波函数对称性受外界环境的影响。然而,这些基函数中不含弥散基,不能很好地描述过渡态和活性中间体弱相互作用和价电子分布的特征,因而该计算水平的基组不合理。在上述计算水平加入了弥散基后(B3LYP/6-311+G*和B3LYP/6-311++G**),过渡态和活性中间体的能量都大于反应物的能量(B3LYP/6-311+G*分别为37.99和9.45 kJ?mol-1,B3LYP/6-311++G**水平分别为40.51和13.01 kJ?mol-1),定性上符合“活性中间体”控制反应的机理和实验推测,因而对像本反应这种含离子反应体系的计算必须在基组中加入弥散基。计算水平最高的M06-2X[11]/def2-TVZPP得到的活化能具有高于反应物的正能量值,符合机理和实验推测;然而该计算水平的中间产物能量高于活性中间体,不符合反应机理。究其原因是M06-2X对电子离域的处理更好,而本反应的分子和离子体系中电子离域效应没有那么强;另外,def2-TVZPP基组包含的弥散成分较小,对反应中的离子描述不准确,故M06-2X/def2-TVZPP计算水平不能适用于本反应。因而,本反应的计算可在B3LYP/6-311+G*和B3LYP/6-311++G**两种水平进行,考虑到计算的经济性,我们采用了B3LYP/6-311+G*水平。
| 图表编号 | XD0056878000 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.06.28 |
| 作者 | 张春芳、张翠妙、李江涛、韩琳玉、顾芳、王海军 |
| 绘制单位 | 河北大学化学与环境科学学院、河北大学化学与环境科学学院、河北大学化学与环境科学学院、河北大学化学与环境科学学院、河北大学化学与环境科学学院、河北大学化学与环境科学学院 |
| 更多格式 | 高清、无水印(增值服务) |




{kind=link}