《表4 miRNA通过作用于其靶点影响IM的敏感性》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
↑药物敏感性增加;↓药物敏感性降低
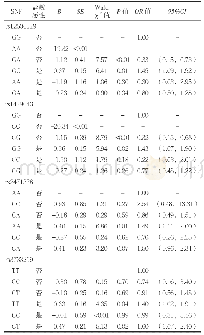
BCR-ABL非依赖性耐药机制在逐步的探索中,如上调Src激酶、ABC转运体介导的药物外排,以及静止干细胞(如原始白血病CD34+CD38-细胞)的存在[10,51]。Jin等[28]发现,K562R细胞的P糖蛋白(PGP)和其编码基因ABCB1的表达均高于K562细胞,miR-214能够与ABCB1 mRNA的3'-UTR结合,转染mi R-214后,ABCB1和PGP均降低,K562R对IM敏感性增加,提示K562R细胞ABCB1 mRNA和PGP表达上调可能与miR-214表达下调有关,mi R-214可能通过调节ABCB1的表达参与CML患者的IM耐药。Dong等[29]证明mi R-328通过降低乳腺癌耐药蛋白(BCRP,基因为ABCG2)的表达,增加K562对IM的敏感性。WNT2在K562R细胞中的表达水平显著升高,其介导的自噬降低CML细胞对IM的敏感性,miR-199a/b-5p直接靶向抑制WNT2表达来减少K562R的自噬,诱导细胞凋亡[31]。信号转导与转录活化因子5(STAT5)被鉴定为miR-221的靶目标,STAT5与miR-221的表达呈负相关,mi R-221通过靶向抑制STAT5诱导CML细胞凋亡和抑制细胞增殖,miR-221上调显著改善K562R对IM的敏感性。此外,当miR-221过表达时,Bax、Caspase3和C-PARP的表达增加,Bcl-2的表达降低[27]。IM耐药CML细胞株的糖酵解升高,mi R-202通过靶向抑制己糖激酶2(HK2)抑制糖酵解,使IM耐药的CML细胞株增敏[32]。IL-8的siRNA及miR-493-5p分别转染K562细胞24 h后,细胞中IL-8均明显下调,转染IL-8 siRNA的K562细胞生长明显抑制且对IM敏感性的升高,转染miR-493-5p的K562细胞生长抑制,但对IM的敏感性未见明显变化,仅能证明miR-493-5p可抑制IL-8的表达,沉默IL-8可抑制细胞生长,增强IM对K562细胞的敏感性,尚不能证明miR-493-5p通过抑制IL-8增强IM的敏感性[21]。Zhou等[30]发现,ST3GAL IV在K562R细胞和CML耐药患者中高表达,ST3GAL IV敲低的K562R细胞多阻滞在G1期且凋亡增加,化疗敏感性恢复。荧光素酶实验证实mi R-224和let-7i直接调控ST3GAL IV基因的表达,上调miR-224或let-7i后ST3GAL IV的表达降低,K562R细胞对IM的敏感性增加。由上推测,mi R-224和let-7i可能通过靶向ST3GAL IV调节白血病细胞的增殖和化疗敏感性。Soltani等[24]通过生物信息学分析确定MYC是miR-451的潜在靶点,IM耐药的CML患者MYC水平升高,猜测miR-451通过调控MYC参与IM耐药。Min等[48]发现,转染Pre-miR-365的K562细胞化疗敏感性和凋亡率均降低,miR-365通过抑制促凋亡蛋白Bax和裂解的Caspase-3在IM敏感的CML细胞中的表达而诱导耐药和减少凋亡;K562R的外泌体处理K562和mi R-365转染的K562后,Bax和Caspase-3的表达均比照组显著下调。K562R细胞的外泌体中mi R-365的表达水平明显高于K562的外泌体,K562R的外泌体可以内化到K562中,并赋予其耐药特性,外泌体可介导miR-365在K562R和K562之间的水平转移。在CML患者中,RanGAP1和miR-1301的表达水平呈负相关,miR-1301可靶向RanGAP1的3′UTR,敲低RanGAP1或增加miR-1301的表达均有利于增加CML细胞对IM的敏感性。此外,研究还证实miR-1301可通过下调RanGAP1的表达,诱导BCR-ABL核包裹和p73反式激活,从而增强IM对CML的疗效[45]。Akt2、STAM2和STAT5A m RNA是miR-2278的功能靶点,转染miR-2278降低了AKT2、STAM2和STAT5A的表达水平,抑制K562细胞增殖,诱导凋亡,提高K562对IM的敏感性[46]。miR-101过表达可抑制K562细胞增殖,促进凋亡:miR-101通过降低STAT5表达促进细胞凋亡;通过抑制NF-κB靶基因使K562细胞对IM增敏;通过降低JAK2表达增强K562细胞对IM的敏感性;通过上调mi R-23a使STAT5、Bcl-2和CCND1表达降低,而mi R-23a可能在STAT5、Bcl-2和CCND1基因的3'-UTR区域存在结合位点;因此,miR-101联合IM治疗CML可能是一种潜在的治疗策略[41]。miR-16的过表达显著降低了MDR1(多药耐药基因1)的m RNA和蛋白水平,长链非编码RNA尿路上皮癌胚抗原1(lncRNA UCA1)作为MDR1的内源竞争RNA(ceRNA),与MDR1 mRNA竞争共同的mi R-16参与CML细胞对IM的耐药[52]。mi RNA-409-5p在儿童CML中低表达,43 kDa核孔蛋白(NUP43)被证明是miRNA-409-5p的靶基因,miRNA-409-5p通过上调NUP43表达使细胞周期阻滞在G0/G1期,并可下调细胞周期蛋白PCNA、C-MYC和cyclin D1的表达,同时上调miRNA-409-5p,增强了IM对CML敏感性[47]。Salati等[49]在白血病干细胞(LSCs)中发现,上调miR-29a-3p和miR-660-5p能够在不影响BCR-ABL激酶活性的情况下,导致它们各自的靶蛋白TET2和EPAS1表达降低,使CML LSCs对TKI产生耐药性;下调CML LSCs中miR-494-3p,能够在不影响BCR-ABL激酶活性的情况下导致其靶蛋白C-MYC的上调,减少TKI诱导的细胞凋亡。沉默miR-21通过阻断PI3K/AKT通路使CML CD34+干(祖)细胞对IM诱导的凋亡敏感[33]。敲低miR-21可使PTEN表达增加,从而使Ph+ALL的Sup-B15细胞对IM增敏[34]。综上分析,在BCR-ABL非依赖性耐药机制的研究中较多涉及ABC转运体、STAT5、c-myc(表4),它们在IM的耐药中发挥着重要的作用。miR-214抑制ABCB1、mi R-328抑制ABCG2,ABCB1和ABCG2均属于ABC转运体的编码基因,ABC转运体过表达可将细胞内药物转运出细胞外,减少细胞内药物浓度,参与细胞耐药[53]。通过上调miR-214和miR-328抑制ABC转运体表达增强细胞内药物浓度,从而达到增加IM敏感性的效果[28-29]。STAT5由STAT5A和STAT5B亚基组成,是酪氨酸激酶癌基因(TKO)的重要下游效应因子,可触发BCR-ABL1突变,促进CML对TKI的耐药[54-55]。上调miR-221、miR-101、miR-2278均可抑制STAT5的表达,增强IM对K562R的细胞毒性[27,41,46]。此外,Nie等[56]发现,STAT5在CML细胞中上调,STAT5通过与pre-mi R-202启动子结合直接激活miR-202-5p转录,USP15是mi R-202-5p的直接靶点,USP15下调导致Caspase-6蛋白水平降低,证明阻断STAT5A/mi R-202-5p/USP15/Caspase-6调节轴能诱导CML细胞凋亡。Xia等[57]认为c-myc是克服AML耐药的潜在治疗靶点,c-myc参与了AML微环境介导的耐药,抑制c-myc可诱导Caspases-3表达,使Bcl-2、Bcl-xl和血管内皮生长因子(VEGF)表达降低,骨髓间充质基质细胞(MSC)可通过c-myc依赖机制保护白血病细胞免于凋亡。miR-451、miRNA-409-5p、miR-494-3p均可影响myc的表达[24,47,49]。ABC转运体、STAT5、c-myc参的与IM耐药具体机制及各蛋白之间的相互作用错综复杂,其完整的网络体系尚未明确。与IM耐药相关的miRNA目前尚处于基础实验阶段,其机制的研究为从根本上解决IM耐药问题提供了方向。
| 图表编号 | XD00170758800 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2020.06.01 |
| 作者 | 曹慧贞、孙航、孙允霄 |
| 绘制单位 | 滨州医学院烟台附属医院儿科、滨州医学院生物化学与分子生物学教研室、滨州医学院烟台附属医院儿科 |
| 更多格式 | 高清、无水印(增值服务) |
![表4 miRNA在MM表观遗传调控中的作用[32]](http://bookimg.mtoou.info/tubiao/gif/LZYX202005013_18300.gif)




{kind=link}