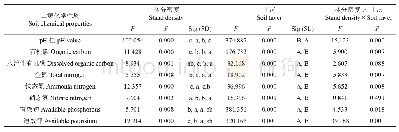
《表1 反应通道主要化学键的电子密度拓扑性质》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
本系列图表出处文件名:随高清版一同展现
《密度泛函理论研究SiFCl自由基和异硫氰酸的反应机理》
反应物中间体IM1生成时,能量降低0.1kJ·mol-1,在IM1中,通过Si、S、C相互作用经TS1形成稳定的三元环中间体IM2,反应势垒为113.8kJ·mol-1。然后IM2经TS2生成产物P1(SC-SiFCNH),反应势垒为113.2kJ·mol-1。在整个反应过程中C-N键长从0.1194nm(IM1)逐渐伸长至0.1295nm(TS2)、0.1382nm(IM2),同时,N-Si键长逐渐缩短,依次从0.3641nm(IM1),0.1901nm(TS1),0.1843nm(IM2),0.1609nm(P2),C-N的削弱有利于C-N键的断裂和C-Si键的生成。由表1所示,N-Si键BCP的ρ(r)逐渐升高至0.1607(P1),表明键强度逐渐增强,直至键生成。2ρ(r)值逐渐增大,表明N-Si键离子性增强。C-N键BCP的ρ(r)从0.4315(IM1)降至0.3024(IM2),表明键强度逐渐减弱,直至断裂生成P1。C-N键的2ρ(r)值逐渐增加,表明键的共价性逐渐减弱,离子性逐渐增强,形象地描述了键的断裂最后生成P1的过程。
| 图表编号 | XD00134857800 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2020.03.01 |
| 作者 | 侯丽杰、韩彦霞、高立国 |
| 绘制单位 | 陇东学院化学化工学院、陇东学院化学化工学院、榆林学院化学化工学院 |
| 更多格式 | 高清、无水印(增值服务) |

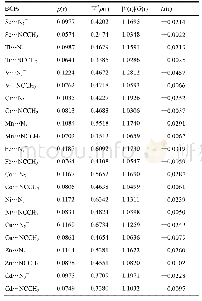
![表5[HAlkyl][TFSA]的主要氢键作用部位BCP处的电子云密度特征](http://bookimg.mtoou.info/tubiao/gif/SHZN202005004_04900.gif)

{kind=link}