《表1 闪锌矿结构金属磷化物的键长、晶胞边长和体积模量》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
注:1=0.1 nm.
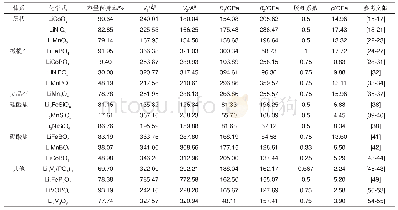
图2为0 K时电子基态能量及零点振动能加和的相对值随晶胞体积的变化,通过能量对体积的一阶导数可以找到晶胞平衡体积及相应的结构性质。其结果总结列于表1,可以看到,B-P、Al-P、Ga-P和In-P键长分别为1.96、2.36、2.34、2.53?,基本呈现一个逐渐增加的趋势,这是因为一方面电负性B>P>Ga>In>Al,另一方面原子半径In>Ga>Al>B,故Ga和P的电负性差虽然较Al更小,即GaP的共价键成分更多,但由于原子半径也更大,两者键长相近;而In和P的电负性差虽然也较Al更小,但是原子半径显著大于Al,所以InP的键长要高于AlP。晶胞边长是固体材料最基本的结构性质之一,并且和键长直接相关。由表1可知BP、AlP、GaP、InP在0 K的晶胞边长分别为4.530、5.449、5.422、5.848?,与化合物键长有着相同变化趋势,总体呈现逐渐增加的变化。此外,通过声子频率的计算可以获得晶胞在不同温度下的Gibbs自由能及此温度下稳定结构,如300 K时BP、AlP、GaP和InP的晶胞边长分别为4.531、5.450、5.427、5.850?。可以看到表1中晶胞边长和体积模量的计算值与实验值及其他理论计算值的一致性很好,这也从侧面说明本文的计算参数设置比较合理。此外,还可以看到晶胞边长会随着温度升高而增大,说明金属磷化物可能属于热膨胀类材料。
| 图表编号 | XD00118112500 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2020.01.01 |
| 作者 | 吴杰、李嘉辉、于燕梅、于养信 |
| 绘制单位 | 清华大学化工系化工热力学实验室、清华大学化工系化工热力学实验室、清华大学化工系化工热力学实验室、清华大学化工系化工热力学实验室 |
| 更多格式 | 高清、无水印(增值服务) |





{kind=link}