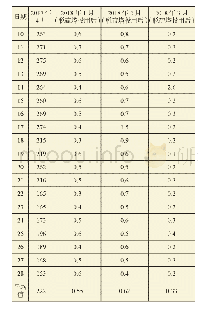
《表5 DFT中BaSn1-xZrxO3的键长(nm)、晶格常数(nm)、体积(nm3)计算结果》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
为验证实验结论的可靠性和合理性,采用基于密度泛函理论的平面波赝势计算程序Cambridge Sequential Total Energy Package(CASTEP)模块中LDA下的Ceperley-Alder-Perdew-Zunger(LDA-CA-PZ)交换关联函数,计算BSZO(x=0,0.5,1.0)的晶胞参数、能带结构及态密度.计算时首先在BSO原胞的基础上建立2×2×2的超晶胞(共40个原子),再对几何模型进行结构优化,其中平面波截止能量为340 eV,布里渊区K矢量选取为3×3×3.优化后的晶格常数、晶胞体积、键长如表5所示.由表5可以看出,Zr—O—Zr键长最长(0.415 3 nm),Sn—O—Sn键长最短(0.407 2 nm),这是因为Zr4+的离子半径大于Sn4+的离子半径此外,计算结果表明,晶格常数与晶胞体积逐渐增大的变化趋势与实验结果相一致.
| 图表编号 | XD00116683000 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.12.10 |
| 作者 | 李凯峰、高强、刘亲壮 |
| 绘制单位 | 淮北师范大学物理与电子信息学院、淮北师范大学物理与电子信息学院、淮北师范大学物理与电子信息学院 |
| 更多格式 | 高清、无水印(增值服务) |

![表3 化合物1的[Mo OBr4(H2O)]-的键长(nm)和键角(°)范围](http://bookimg.mtoou.info/tubiao/gif/WJHX202102014_02500.gif)


{kind=link}