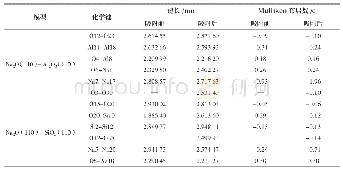
《表2 最优结构和612℃动力学模拟后结构中O-H键的键长和布居数》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
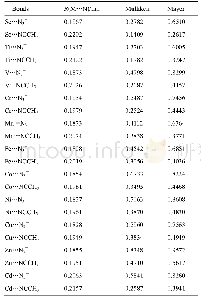
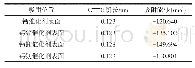
根据试样的TG-DSC曲线可知,蛇纹石在612℃时发生了脱羟基相转变过程。初始态的Mg3Si2O5(OH)4超晶胞经过612℃动力学模拟计算后,相应计算参数见表2—表4。表2为最优结构和动力学模拟后结构中O-H键的键长和布居数。表3为最优结构和612℃动力学模拟后结构中镁离子间距。表4为最优结构和动力学模拟后结构中Si-O键的键长和布居数。图4为动力学模拟前后的Mg3Si2O5(OH)4构型。
| 图表编号 | XD0029123600 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.04.25 |
| 作者 | 王宇鲲、魏永刚、彭博、李博、周世伟 |
| 绘制单位 | 昆明理工大学省部共建复杂有色金属资源清洁利用国家重点实验室、昆明理工大学冶金与能源工程学院、昆明理工大学省部共建复杂有色金属资源清洁利用国家重点实验室、昆明理工大学冶金与能源工程学院、昆明理工大学省部共建复杂有色金属资源清洁利用国家重点实验室、昆明理工大学冶金与能源工程学院、昆明理工大学省部共建复杂有色金属资源清洁利用国家重点实验室、昆明理工大学冶金与能源工程学院、昆明理工大学省部共建复杂有色金属资源清洁利用国家重点实验室、昆明理工大学冶金与能源工程学院 |
| 更多格式 | 高清、无水印(增值服务) |



{kind=link}