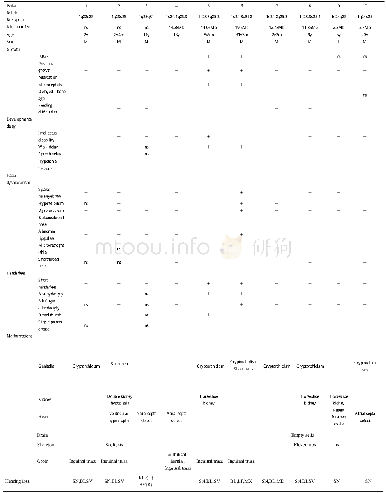
《表1 1q22-q25.2区缺失患者听力损失的临床特征》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
本系列图表出处文件名:随高清版一同展现
《一例1q23.3-q25.2缺失综合征型耳聋报道及文献复习》
由于aCGH技术对全基因组进行扫描,可以检测染色体内微小缺失、重复等不平衡性的重排,国际细胞基因组芯片标准协作组(International Standards for Cytogenomic Arrays Consortium,ISCA Consortium)推荐将aCGH作为对原因不明的发育迟缓、智力低下、多种体征畸形以及自闭症患者的临床首选检测方法[6]。本文通过aCGH技术检测患儿为1q23.3-q25.2区17.67 Mb杂合缺失,属于1q近端缺失,由于患者父母无相同片段染色体异常,提示该患儿1q长臂缺失是自发突变引起。对于1q区域缺失引起的听力下降,早在1996年Fagerheim等[7]首次报道了DFNA7基因(OMIM:601412),定位于1q21-q23,为常染色体显性遗传,临床特征以高频开始,渐进性感音神经性聋。随后,2009年Catelani等[8]研究表明1q23.3-q25.2片段可能含有与听力相关的剂量敏感基因,且DFNM1基因(OMIM:605429)位于1q24已证实与听力障碍有关[9]。文献检索发现有10例报道1q22-q25.2区缺失的患者存在听力损失,这些区域的缺失与1q23.3-q25.2部分重叠,在一定程度上可以解释该患儿听力损失的原因(表1)。
| 图表编号 | XD00114140000 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.10.15 |
| 作者 | 张飞、牛文侠、丁韶洸、谢存存、贾晓东、秦利涛、刘宏建 |
| 绘制单位 | 郑州大学附属儿童医院河南省儿童医院郑州儿童医院耳鼻咽喉科、河南大学人民医院、河南省人民医院耳鼻咽喉科郑州大学人民医院河南大学人民医院、河南省人民医院耳鼻咽喉科郑州大学人民医院河南大学人民医院、河南省人民医院耳鼻咽喉科郑州大学人民医院河南大学人民医院、郑州大学人民医院河南省人民医院河南省医学遗传研究所、河南省人民医院耳鼻咽喉科郑州大学人民医院河南大学人民医院 |
| 更多格式 | 高清、无水印(增值服务) |



{kind=link}