《表1 优化参数:过渡金属的能带结构与声子谱的第一性原理计算》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
基于第一性原理的密度泛函理论(DFT),密度泛函微扰理论(DFPT),本文利用晶体结构计算程序包abinit[7],采用广义梯度近似(GGA),PerdewBurke-Ernzerhof(PBE)泛函[8]和Troullier-Martins(TM)赝势完成相应的第一性原理计算,计算时引入了电子的自旋极化。为了保证计算过程中能量得到有效收敛,分别优化计算得到平面波的截断能E-cut和k点的数量如表1所示。此外为了确保能量随晶格参数变化的连续性,特引入了光滑拖尾参数smearing和晶格参数的最大缩放比dilatmx。
| 图表编号 | XD0045253600 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.04.18 |
| 作者 | 陈春彩、陈源福 |
| 绘制单位 | 闽南理工学院物理教研室、闽南理工学院物理教研室 |
| 更多格式 | 高清、无水印(增值服务) |
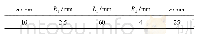
![表4 Al、Mg和Zn的热力学参数计算值与实测值[7,28,30~33]与声子热导率](http://bookimg.mtoou.info/tubiao/gif/JSXB202103011_05900.gif)

![表1 EAM势函数中的Fe和Cu表面能γ,与第一性原理(FP)[7]计算和实验(Exp)[8]测试表面能的比较](http://bookimg.mtoou.info/tubiao/gif/GCHZ202004008_01000.gif)


{kind=link}