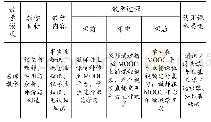
《表3 BS的遗传学分类及其特点》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
注:NKCC2:噻嗪类敏感的Na-K-2C1协同转运蛋白;ROMK:内流性电压依从性K+通道;C1C-Ka:氯离子通道-Ka;C1C-Kb:氯离子通道-Kb;barttin蛋白:ClC-Ka和ClC-Kb通道β亚基;CaSR:细胞外钙敏感受体;TAL:髓袢升支粗段;DCT:远曲小管;CCD:中心集合小管;AR:常染色体隐性遗传;AD常染色体显性遗传
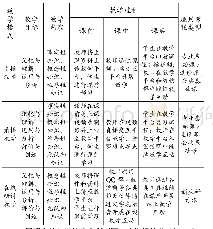
目前BS诊断依据尚无统一标准,但具备以下几点可作出临床诊断:(1)长期低钾血症,慢性肾性失钾(血钾<2.5mmol/L,尿钾能检测到;血钾2.5~3.0mmol/L,尿钾>20mmol/24h;血钾3.0~3.5mmol/L,尿钾>25mmol/24h);(2)低氯性代谢性碱中毒;(3)RAAS系统激活;(4)血压正常;(5)前列腺素增多,肾脏病理提示球旁器细胞增生,此项不是诊断必备条件;(6)确诊依据:基因检测。BS为常染色体遗传性失盐性肾小管疾病,为明确BS诊断,抽该患者及其父亲外周血外送基因检测,其结果提示与BS相关的基因病变有CLCNKA和KCNJ1(表3),CLCNKA基因是BS IVb型的致病基因,检测到患者的CLCNKA基因位于Exon8的错义突变c.747A>G(p.Ile249Met),为杂合变异,蛋白功能损伤预测结果提示有可能致病。BS IVb型是CLCNKA和CLCNKB共同病变所致,目前国内外对BSIVb型报道极少,仅检索到2例病例报道[4-5]。本例患者未检测到CLC-NKB病变,而单纯的CLCNKA错义杂合突变是否会引起临床症状尚不确定,因此该变异致病性有待进一步研究。KCNJ1基因是BS II型的致病基因,检测到KCNJ1基因位于Exon2的错义突变c.625C>T(p.Leu209Phe),蛋白功能损伤预测结果为有可能致病。KCNJ1基因突变,使ROMK通道活性降低,细胞内钾离子转出细胞受限,为NKCC2提供的K+减少,导致K+、Na+、CL-转运发生障碍,Ca2+被动吸收减少,从而引起一系列临床症状,如低血钾、尿钾增多、血容量相对不足、RAAS激活、前列腺素增多、以及球旁器细胞增生、尿钙排除增多及肾钙化等。BS II型大多在新生儿期发病,因此临床分型为新生儿型BS。目前迟发性II型BS也有相关报道[6-7],但成人发病极其罕见。检索相关文献及人类基因突变数据库,以上两个位点的基因突变暂时没有相关报道,有可能是BS基因突变的新发位点。另外患者还检测到PRX基因位于Exon7的错义突变c.3673G>A(p.Val1225Met),为杂合变异。PRX基因定位在19q13.1-19q13.3,编码保护周围神经髓磷脂的蛋白质,是腓骨肌萎缩症4F型与Dejerine-Sottas病的致病基因,其主要临床表现为四肢远端进行性肌无力及肌萎缩,感觉丧失显著。该患者检测到的PRX基因变异的致病性已有相关文献报道[8]。但是该患者仅有运动发育迟缓、乏力,无行走困难、肌肉萎缩、高弓足、足下垂、爪形手、脊椎畸形、感觉性共济失调等表现,未行神经电生理及神经活检等相关检查,不能确定是否存在腓骨肌萎缩症4F型和Dejerine-Sottas病。患者父亲的CLCNKA及KCNJ1基因表现与患者的变异表现一致,PRX基因未发现变异。但患者父亲未出现BS相关临床表现,这可能与其父亲基因突变效应尚不能引起临床症状有关。
| 图表编号 | XD0037520300 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.04.20 |
| 作者 | 代敏、李飞、李思成、杨波、李显文 |
| 绘制单位 | 遵义医学院及其附属医院内分泌科、遵义医学院及其附属医院内分泌科、遵义医学院及其附属医院内分泌科、遵义医学院及其附属医院内分泌科、遵义医学院及其附属医院内分泌科 |
| 更多格式 | 高清、无水印(增值服务) |


{kind=link}