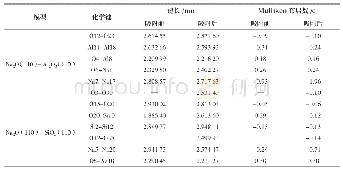
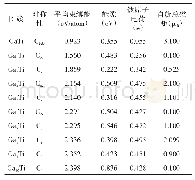
《表2 GeLi62+·nH2团簇Li—Ge, H2—Li, H—H的平均键长和平均吸附能以及自然电荷分布》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
氢分子在GeLi62+表面吸附的结构特征、平均吸附能、前线轨道能隙及H2、Li和Ge原子的自然电荷分布列于表2,Ge—Li平均键长、Li与H2平均距离随吸附氢分子数目增多而增大。氢分子平均吸附能在2.145~4.004 kCal.mol-1之间,介于物理吸附和化学解离吸附之间。表2还可见在每个Li原子周围吸附1~3个H2时,氢分子的平均吸附能随吸附氢分子数目增多而减小。这可以从自然电荷分布来加以分析,表2可见,随吸附氢分子增加,氢分子失电子数量有少量增加,随即转移到Li+上,另外还可见,中心Ge离子上的负电荷也随吸附氢分子的增加而减少,这可以理解为Ge离子上的负电荷转移到了Li+上发生了正负电荷的中和,导致GeLi62+体系产生的静电场下降,从而降低了与氢分子之间的静电相互作用,当吸附到四H2时相互作用显著变弱。再者由于GeLi62+表面周围空间有限,氢分子继续增加,空间出现位阻效应,导致不能有效吸附更多H2,再次说明体系吸氢具有饱和性。
| 图表编号 | XD0034859600 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.01.01 |
| 作者 | 阮文、冯五强、温在国、陆彪、吴永波、陈银坤 |
| 绘制单位 | 井冈山大学数理学院、井冈山大学数理学院、井冈山大学数理学院、井冈山大学数理学院、井冈山大学数理学院、井冈山大学数理学院 |
| 更多格式 | 高清、无水印(增值服务) |




{kind=link}