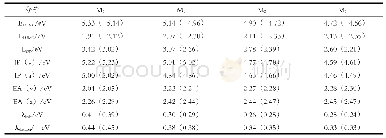
《表3 在TD-B3LYP(TD-PBE0)/6-31+G(d)水平上化合物M0~M3最主要的电子吸收光谱数据》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
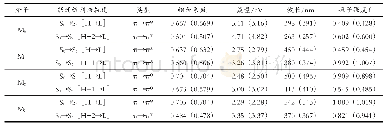
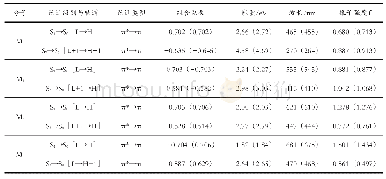
M0~M3化合物的主要吸收光谱数据列于表3,绘制模拟吸收与荧光光谱于图3.图3清楚表明,增加吡咯环,最大吸收波长明显红移;而表3数据则表明S0→S1跃迁的振子强度随吡咯环增加而显著增大.在TD-B3LYP(TD-PBE0)水平上和甲醇溶剂中,预测M0~M3最大吸收波长分别为263(257)、380(374)、500(496)和542(543)nm,见图3;预测M0~M3分子的S0→S1跃迁吸收波长分别为398(391)、450(444)、500(496)和542(543)nm,见表3.分子轨道分析表明,S0→S1跃迁均为HOMO→LUMO之间的π→π*跃迁.S0→S1跃迁振子强度f≥0.409(0.428),所以HOMO→LUMO均为较强吸收.值得指出的是,对于M0,f7>f1,故S0→S7是最强吸收.对于M1,f3>f1,故S0→S3是最强吸收.而对于M2、M3,S0→S1是最强吸收.
| 图表编号 | XD00161293600 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2020.06.01 |
| 作者 | 庾弘朗、才红、朱勋、薛晓婷 |
| 绘制单位 | 韩山师范学院化学与环境工程学院、韩山师范学院化学与环境工程学院、韩山师范学院材料科学与工程学院、韩山师范学院化学与环境工程学院 |
| 更多格式 | 高清、无水印(增值服务) |
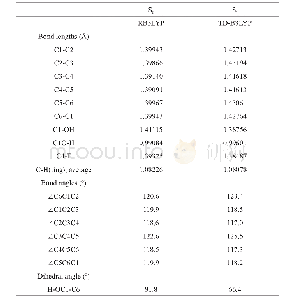
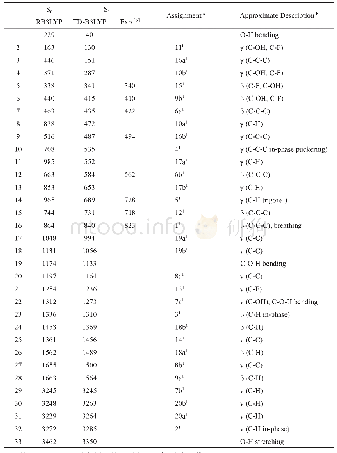
![表1 在[B3LYP(PBE0)//TD-B3LYP(TD-PBE0)]/6-31+G(d)理论水平上计算的化合物M0~M3偏离标准平面数据](http://bookimg.mtoou.info/tubiao/gif/HSSC202003007_01600.gif)
{kind=link}