《表2 噻唑环上不同取代基的化合物的抗结核活性》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
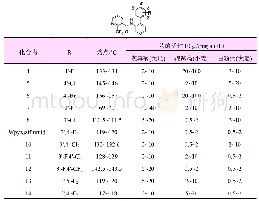
以Ciprofloxacin(环丙沙星)为阳性对照,测试了合成的19个化合物在1和0.1μmol/L浓度下,对结核分枝杆菌(H37Ra)的抑制率(Inh/%)。首先测试了化合物3的活性,在1μmol/L浓度下,抑制率为99.1%,在0.1μmol/L浓度下,抑制率为-11.8%,MIC=0.16μmol/L(如图1)。然后,测试接上噻唑环的新结构化合物,如表2所示。最初发现,噻唑4-位上分别接甲基、叔丁基和苯基的化合物5a、5b和6a,在1和0.1μmol/L浓度下,均未表现出活性。但是,在苯环的对位接上Cl取代,得到化合物6b,在1μmol/L浓度下,抑制率为100.03%。进一步改变Cl取代基的位置,发现间位取代的6c和邻位取代的6d在1μmol/L浓度下活性下降,其中邻位取代的化合物6d活性消失。因为化合物6b的活性较好,在化合物6b的基础上,将苯环对位的Cl替换为Br,得到化合物6e,1μmol/L抑制率为99.1%,0.1μmol/L抑制率为62.6%,活性略低于化合物6b(0.1μmol/L,Inh=80.7%)。与此同时,在苯环上的不同取代位置引入—CF3、—NO2、—CH3和—OCH3,对比它们的抑制率可知,—CF3对位取代的化合物6f活性较好,在1和0.1μmol/L浓度下,抑制率分别达到99.6%和93.4%,而—CF3邻位取代的化合物6g活性消失。—NO2对位取代的化合物6h比间位取代的化合物6i活性高,0.1μmol/L浓度下,化合物6h保持64.2%的抑制率,化合物6i无活性;—CH3取代的化合物,只有对位取代的化合物6j在1μmol/L浓度下,保持90.2%的抑制率,间位6k活性消失;—OCH3对、间位取代的化合物6l和6m抑制率均较低,只有甲氧基二取代的化合物6n活性较好,在1μmol/L浓度下,抑制率为95.0%。
| 图表编号 | XD00126786600 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2020.02.10 |
| 作者 | 何冰、钟鑫鑫、冉凯、奉强、余登斌、韩涛、李仲辉、余洛汀 |
| 绘制单位 | 成都师范学院化学与生命科学院、功能分子研究所、成都师范学院化学与生命科学院、功能分子研究所、四川大学华西医院生物治疗国家重点实验室、生物治疗协同创新中心、四川大学华西医院生物治疗国家重点实验室、生物治疗协同创新中心、成都师范学院化学与生命科学院、功能分子研究所、中国科学院长春应用化学研究所电分析化学国家重点实验室、成都师范学院化学与生命科学院、功能分子研究所、成都师范学院化学与生命科学院、功能分子研究所、四川大学华西医院生物治疗国家重点实验室、生物治疗协同创新中心 |
| 更多格式 | 高清、无水印(增值服务) |





{kind=link}