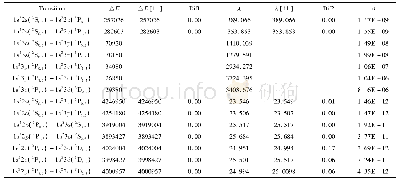
《表1 化合物3a和3b最低光谱跃迁S1和T1的计算波长和主要贡献轨道》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
本系列图表出处文件名:随高清版一同展现
《基于咔唑衍生物的热激活延迟荧光材料合成与性质研究》
图5为理论计算模拟化合物3a和3b的紫外吸收光谱,并与实验光谱做了比较.表1总结了含时密度泛函理论(TDDFT)计算的主要电子跃迁情况.从紫外吸收光谱的模拟图(见图5)可以看出,理论计算结果与实验数据基本吻合.化合物3a和3b在最低能量的电子跃迁,即363和349nm处的吸收峰,分别来自于HOMO→LUMO和HOMO-1→LUMO的轨道跃迁.即最低单重态能量跃迁(S1)来源于分子内电荷转移(ICT,intramolecular charge transfer).同时我们通过TDDFT计算了2个化合物的最低三线态能量跃迁(T1)情况,结果表明都来源于HOMO-1→LUMO的电子跃迁,同样具有ICT特征.
| 图表编号 | XD00116741300 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.12.01 |
| 作者 | 张倩、袁丁、刘凯、左梓岑、李高楠、钮智刚 |
| 绘制单位 | 海南师范大学化学与化工学院、海南师范大学化学与化工学院、海南师范大学化学与化工学院、海南师范大学化学与化工学院、海南师范大学化学与化工学院、海南师范大学化学与化工学院 |
| 更多格式 | 高清、无水印(增值服务) |





{kind=link}