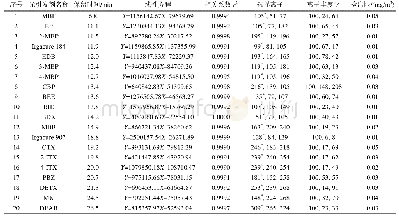
《表2 光引发剂C2P及C2V的理论计算数据》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
如表2所示,香豆素肟酯光引发剂C2V及C2P的第一激发态S1为HOMO-LUMO跃迁(属于π-π*跃迁),能量均较低(等效400nm波长光激发),且均具有较大的振子强度f(fC2V为1.6676,fC2P为1.6575),表明该类分子在可见光区具有强烈吸收,此结果与紫外-可见吸收光谱相符(图3a)。但两种引发剂的第二激发态S2与第三激发态S3能量均较高(属于n-π*跃迁,等效300nm波长光激发),且振子强度较低,对可见光区域吸收较弱。肟酯类分子受到激发后会发生N—O键断裂,并通过脱羧产生自由基以引发聚合。两种引发剂在HOMO-1和HOMO轨道跃迁至LUMO轨道过程中,主要发生杂原子上n电子和共轭结构的π电子向共轭结构的π*轨道转移,向肟酯基团的σ*轨道转移概率较低(图4)。对比C2P与C2V的LUMO轨道中肟酯基团σ*电子云的分布,C2V分子发生n-σ*和π-σ*跃迁概率更高,理论上具有更快的断键速率和更高的反应速率,这与稳态光解(图6)及单光子聚合动力学结果(图7)相符。
| 图表编号 | XD0087606000 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.09.01 |
| 作者 | 李梦琦、邱婉婉、胡鹏、朱俊哲、刘仁、李治全 |
| 绘制单位 | 江南大学光响应功能分子材料国际联合研究中心、江南大学化学与材料工程学院、江南大学化学与材料工程学院、江南大学化学与材料工程学院、江南大学化学与材料工程学院、江南大学光响应功能分子材料国际联合研究中心、江南大学化学与材料工程学院、江南大学光响应功能分子材料国际联合研究中心、江南大学化学与材料工程学院 |
| 更多格式 | 高清、无水印(增值服务) |




{kind=link}