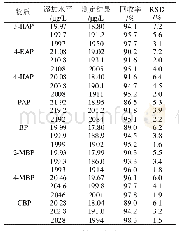
《表2 光引发剂2-OAME和4-OAME的光学数据》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
本系列图表出处文件名:随高清版一同展现
《香豆素并咔唑基草酸酯类UV-LED可激发光引发剂的合成与研究》
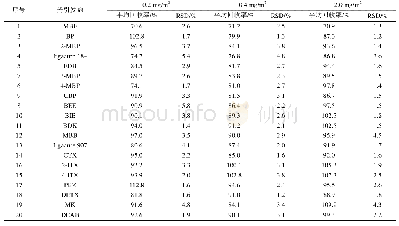
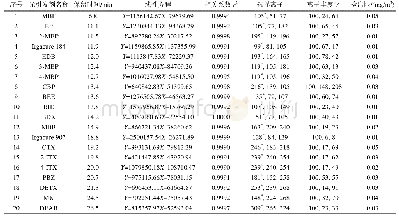
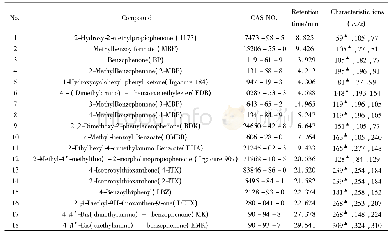
两个光引发剂分子在紫外光区都有较大吸收且吸收光谱范围均延伸至可见光区,其中4-OAME的最大吸收波长λabs=373nm(摩尔消光系数εmax~14300L·mol-1·cm-1)[20],相较于2-OAME(λabs=366nm,εmax~10600L·mol-1·cm-1)更红移,二者均与商业化的LED光源的发射光谱重合较好,特别是在365~405nm下表现出较好的光吸收性质,在各个波长下的摩尔消光系数如表2所示,4-OAME在385nm处依然有7400L·mol-1·cm-1。图5为两个分子利用理论计算优化后的分子结构,可以明显观察到咔唑并香豆素的共轭主体结构部分具有非常好的平面性,π-电子在整个共轭体系中可以发生离域,这是两个分子在近紫外-可见光区有较大吸收的根本原因。但两个同分异构体的平面构型不同是分子之间吸收差异的来源,2-OAME分子基本成线性,而4-OAME分子共轭体系成直角[20]。两个OAME分子的前线轨道的电子云分布和能量最低的两个跃迁的计算结果如图6所示,具体数据见表1。从中可以发现S0→S1涉及到的主要是羰基上氧的孤对电子的n→π*跃迁,S0→S2主要是HOMO轨道到LUMO轨道的π→π*跃迁,特别是2-OAME的理论计算的振子强度分布和实际吸收波谱的吸收峰值有很高的吻合度(图4),进一步说明对其前线轨道推测的性质[25]。虽然两个分子构型不同,当时跃迁的本质是相同的。
| 图表编号 | XD0087605300 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.09.01 |
| 作者 | 周茹春、柳宜卓、陈世雄、王伟杰、陈思琦、金明 |
| 绘制单位 | 同济大学材料科学与工程学院高分子系、同济大学材料科学与工程学院高分子系、同济大学材料科学与工程学院高分子系、同济大学材料科学与工程学院高分子系、同济大学材料科学与工程学院高分子系、同济大学材料科学与工程学院高分子系 |
| 更多格式 | 高清、无水印(增值服务) |


{kind=link}