《表2 不同S掺杂浓度Sn Ti O3-xSx (x=0, 0.33, 0.5, 1) 的键长及其垂直距离z的差值 (NV表示最近邻垂直的位置, NP为最近邻平行的位置)》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
/nm
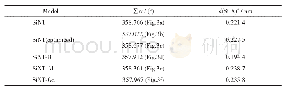
计算得到SnTiO3的晶格常数为a=0.380 nm,c/a的值为1.17,与之前的计算结果(a=0.385 nm,c/a=1.13)[20]相吻合。SnTiO3与PbTiO3相比[12],具有更大的c/a值,有利于获得大的极化强度。如表1所示,当S掺入到SnTiO3后,SnTiO3-xSx(x=0,0.33,0.5,1)仍然保持P4mm空间群,与SnTiO3一样具有四方结构。SnTiO3-xSx的晶格常数a与SnTiO3相比几乎不变,但晶格常数c发生明显的增大。当x=1时,c/a的值由SnTiO3的1.17增加到1.45(表1)。从图1可以看出,晶格常数c方向的变化主要来源于S所在八面体晶格的拉伸。例如,在x=0.5时,S所在八面体的c=0.541 nm,而没有S的Ti-O八面体的c=0.451 nm。可以看出,S掺入后主要改变S所在八面体的结构。c/a值的显著增大的现象在S掺杂PbTiO3上也被观察到[12]。在实验上,S替代O后能够引起金属氧化物的c/a值增大[21-22],形成-S-M-O-M-S-的链状模式,这与我们的结果是一致的。S的掺入会引起SnTiO3原有阴阳离子的成键键长变化。由表2可以看出,Sn-ONV(图1a中Site1位置的O)键长和在c方向垂直距离随掺杂S的增加变化不明显,但要比没有掺杂时(即SnTiO3)减小0.007 nm。Sn-S的键长在x=0.33和0.5时保持不变,在x=1时键长减少0.003 nm,c方向垂直距离减少0.005 nm。Ti-ONP键长在x=0.33和0.5时几乎保持不变,垂直距离也相差较小,但x=1时,Ti-ONP的垂直距离变化相对比较明显,比SnTiO3时的Ti-ONP减少0.011 nm。Ti-ONV和Ti-S键长几乎保持不变。综上所述,当x=0.33和0.5时,只有SnONV在c方向垂直距离要比未掺杂时有一些减少外,其它阴阳离子的键长变化都不是很明显,这说明当S掺杂量不大的时候对离子成键影响比较小。但当x=1时,Sn-S和Ti-ONP在c垂直距离都变化较明显,这说明较大的S掺杂对其离子成键有明显的影响,这也许是SnTiO2S具有金属性的原因。
| 图表编号 | XD0072984100 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.06.01 |
| 作者 | 魏丽静、郭建新 |
| 绘制单位 | 华北电力大学科技学院、河北大学物理科学与技术学院 |
| 更多格式 | 高清、无水印(增值服务) |




![表1 Ti3SiC2各原子之间的键长和键角[11]](http://bookimg.mtoou.info/tubiao/gif/BMJS202001018_00900.gif)
{kind=link}