《表4 αS研究高频共被引文献》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
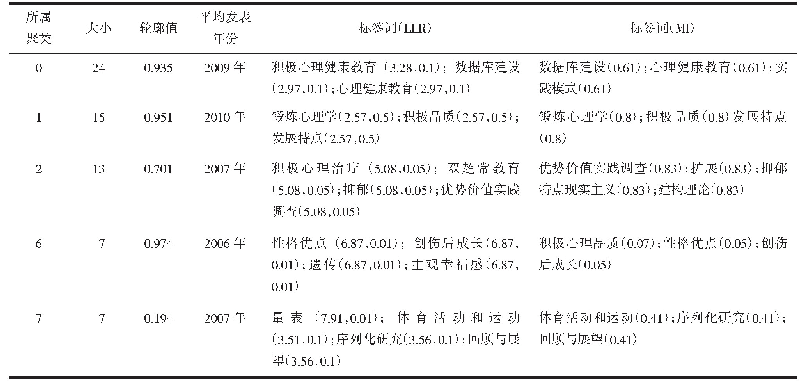
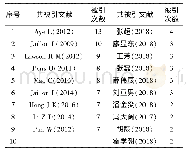
本研究对近20年αS相关文献的被引文献进行共被引分析,重点分析、探测高频共被引文献的主要研究方向,以更好地发现αS研究领域的知识基础和发展脉络。表4显示出αS研究前十篇高频共被引文献,这10篇文献均是αS研究领域的经典文献。1997年,Spillantini等[3]通过免疫组织化学实验首次发现PD患者脑组织中大量不溶性包涵体的主要成分是αS。随后,他们利用免疫电镜发现路易小体及其突触会被指向αS氨基末端和羧基末端序列的抗体强烈染色,能显示出全长或接近全长αS的存在,而且αS染色的结构数量超过对泛素免疫反应的结构数量,从此αS染色取代泛素染色成为检测路易体及其神经突触的首选方法[11]。Baba等[12]从路易体痴呆患者大脑中提纯出路易体进行光和电子显微的免疫细胞化学研究,结果显示αS而非β-突触核蛋白,是散发性PD和路易体痴呆中路易小体的主要成分。引用量排名第1、3、6的文献分别介绍了αS基因的3种错义突变。1997年,Polymeropoulos等在常染色体显性遗传性PD家系中首次发现了αS-A53T突变(第53位丙氨酸突变为苏氨酸),该突变是由αS基因209位的鸟嘌呤被腺嘌呤取代(G209A)引起的;他们预测A53T突变将导致αS的α螺旋被破坏,形成易于聚集的β片层结构[13]。1998年,Kruger等[14]在一个德国PD家系中发现αS基因的第3号外显子中第88位鸟嘌呤被胞嘧啶取代(G88C),从而导致αS-A30P突变(第30位丙氨酸突变为脯氨酸)。2004年,Moore等在一个伴随不同程度的痴呆和视幻觉的西班牙PD家系中检测出突变体E46K(第46位谷氨酰胺突变为赖氨酸)。与野生型及A53T、A30P突变相比,E46K突变明显增加了与带负电荷脂质体的结合能力,并增加了丝状纤维的聚集能力[15]。除了αS基因突变,αS过表达也可导致家族性PD。Masliah等[16]对表达野生型人类αS的转基因小鼠进行研究发现,人类αS的神经元表达会导致某些内含物在新皮层、海马和黑质神经元中不断累积,野生型αS的积累可能是引发PD和相关病症的原因。Singleton等[17]对一常染体显性遗传家系进行研究,发现αS基因的二倍体及三倍体就能导致常染色体显性遗传性PD的发生,而此时αS基因并没有发生突变,表明正常αS基因表达水平的提高可能导致PD。除了αS基因突变,还有许多其他与家族性PD关联的基因突变,如Parkin、PINK1、UCHL1、DJ-1等。1998年,Kitada等[18]在一个日本青少年型PD家系中定位并克隆了Parkin基因,同时发现Parkin基因突变可导致常染色体隐性遗传的青少年型PD。随着对PD患者尸检的增加,科研人员发现PD患者神经系统的病理受累情况以及病理改变错综复杂,统一的病理发展体系亟待提出。2003年,Braak等依据发病进程将PD病变分成6个阶段,即帕金森病Braak分期,该分期可以很好地解释PD的运动症状和非运动症状的演变,在近年来得到了大量的应用,为PD预防、早期诊断、病理生理机制、神经保护等方面的研究提供了理论基础[19]。
| 图表编号 | XD0048763200 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.04.28 |
| 作者 | 邹昱、闾坚强、张庆文、倪鑫、陈佩杰、钱振宇 |
| 绘制单位 | 上海体育学院体育教育训练学院、上海体育学院运动科学学院、上海体育学院体育教育训练学院、第二军医大学基础部生理学教研室、上海体育学院运动科学学院、上海体育学院运动科学学院 |
| 更多格式 | 高清、无水印(增值服务) |




{kind=link}