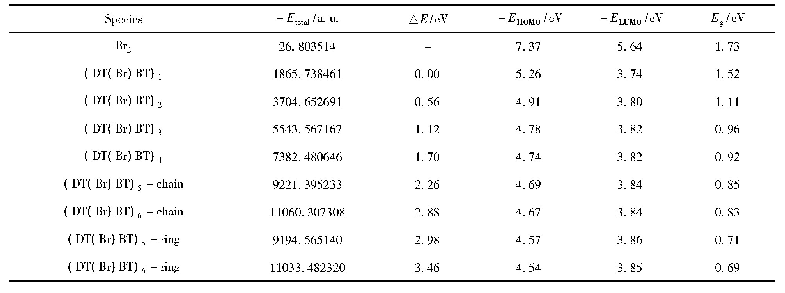
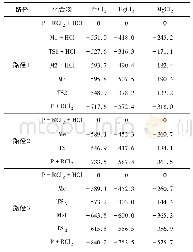
《表3 迪美唑在TiO2 (101) 晶面上反应各驻点的能量E、相对能量Erel、频率v》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
本系列图表出处文件名:随高清版一同展现
《迪美唑在光催化剂TiO_2表面吸附特征及降解机理的理论研究》
在TiO2(101)晶面上,反应通道I的机理为Re→TS1→M1→TS2→P,在Re中,咪唑环上的N(3)原子与TiO2(101)表面Ti(5)原子之间的距离为2.602?TiO2表面上的羟基自由基进攻迪美唑上C(2)原子C(2)–O(1)键长为1.408?,C(2)–N(1)键长由原来的1.370?增加到1.499?,N(3)–C(2)键长由原来的1.343?拉长到1.488?,C(2)–N(1)键、N(3)–C(2)键比其咪唑环上的其他共价键都明显增长,C(2)–N(1)键又比N(3)–C(2)键稍长,所以C(2)–N(1)键断裂开环的可能性最大.在过渡态TS1中,N(1)–C(2)键开始断裂,键长拉长到2.2 2 5?,该步骤R e→T S 1的活化能为25.96 kcal/mol,过渡态有唯一虚频为496.6 i cm-1,根据过渡态的判据此过渡态是真实的(表3).在M1中,N(1)–C(2)键完全断裂,二者之间的距离为3.048?,在C(2)原子处形成烯醇式结构.在TS2中,H(1)原子在O(1)和N(3)原子迁移,O(1)与H(1)和H(1)与N(3)原子之间的距离分别为1.831和1.963?,N(3)从TiO2(101)表面Ti(5)原子上脱离,C(2)原子烯醇式结构上的H(1)向N(3)原子转移,形成四元环过渡态.由于O(1)和N(3)与TiO2(101)表面Ti(5)的距离分别为3.056和2.292?,表明过渡态TS2离TiO2表面较远,二者之间的吸引力较弱,导致此过程活化能高达96.14 kcal/mol,该步反应成为反应通道I的速控步骤.从活化能上判断,迪美唑在TiO2(101)晶面上以反应通道I进行开环反应发生可能性小.
| 图表编号 | XD0034640500 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.02.20 |
| 作者 | 徐伯华、张福兰、魏维、陈晓、李来才、田安民 |
| 绘制单位 | 长江师范学院化学化工学院、长江师范学院化学化工学院、四川师范大学化学与材料科学学院、四川师范大学化学与材料科学学院、四川师范大学化学与材料科学学院、四川大学化学学院 |
| 更多格式 | 高清、无水印(增值服务) |




{kind=link}