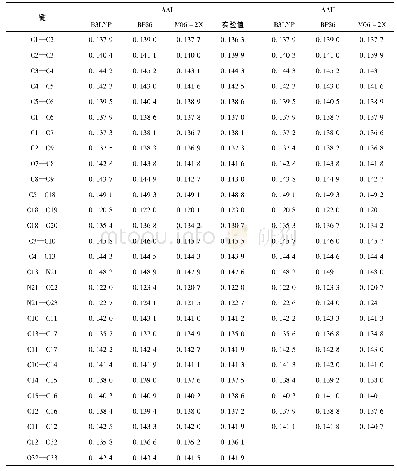
《表1 基于Ga/ZSM-5 (3T簇) 用UB3LYP和UM06-2x方法计算的反应路径中各驻点的相对能量ΔEelec对照》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
本系列图表出处文件名:随高清版一同展现
《N_2O和CO在Ga/ZSM-5上吸附和反应机理的密度泛函研究》
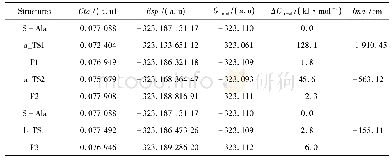
aAbsolute energy E=-3 348.098 113 au.bAbsolute energy H=-3 347.730 629 au.
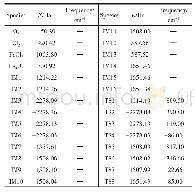
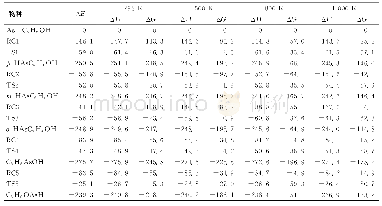
由于分子筛体系较大,簇模型方法是用量子化学方法来研究分子筛的常用方法之一[19-20].本文中采用3T阳离子簇模型(如图1所示)模拟Ga/ZSM-5分子筛的活性中心,以研究CO和N2O在其中吸附和反应的机理.具体来说,在ZSM-5分子筛骨架中截取含有2个Si四面体和1个Al四面体的3T簇[H3Si OAl(OH)2OSi H3],将Ga置于2个O原子的桥位,截取簇时产生的悬挂键用H饱和(以下简记Ga+-Z (3T)) .因分子筛中的吸附和催化反应主要集中在含硅铝桥羟基的特定环境中进行,这种简化的小尺寸的模型簇,既抓住了分子筛中反应的关键,又使理论计算成为可能.类似的处理方式成功见于众多的文献报道[19-20,26-27].用自旋非限制性的杂化密度泛函UB3LYP[28-30]理论方法、6-31+G*基组对吸附复合物、反应的二重态势能面上所有驻点进行几何优化、振动分析,对过渡态做IRC计算.所选泛函已成功用于主族离子交换分子筛的计算,能得到可靠结果[17,26-27].为了验证所选模型及方法对本文体系的适用性,继续做了若干标定计算.首先,对反应所涉及到的各物种,在UB3LYP优化几何基础上用UM06-2x[31]方法做单点能计算,与UB3LYP计算结果一起列于表1.表1数据显示,除UM06-2x方法计算的能垒稍大外,两种方法所得势能曲线趋势是一致的.另用UB3LYP方法及简化模型计算得到反应的第一步中N2O的η1-O吸附能为-67.7 k J/mol与用体现Si O4结构的[(HO)3Si OAl(OH)2OSi(OH)3]簇及UM06-2x方法计算的-53.5 k J/mol相差较小.这些说明,所选模型及计算方法对所研究的反应体系基本适用.所有量化计算均采用Gaussian 09程序[32]完成.计算的吸附能为吸附过程前后的焓变:
| 图表编号 | XD0024702800 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2018.03.05 |
| 作者 | 郭玥、刘琼、张干兵、钟欣欣、胡玮、田丽红、夏清华 |
| 绘制单位 | 有机化工新材料湖北省协同创新中心有机功能分子合成与应用教育部重点实验室湖北大学化学化工学院、有机化工新材料湖北省协同创新中心有机功能分子合成与应用教育部重点实验室湖北大学化学化工学院、有机化工新材料湖北省协同创新中心有机功能分子合成与应用教育部重点实验室湖北大学化学化工学院、有机化工新材料湖北省协同创新中心有机功能分子合成与应用教育部重点实验室湖北大学化学化工学院、有机化工新材料湖北省协同创新中心有机功能分子合成与应用教育部重点实验室湖北大学化学化工学院、有机化工新材料湖北省协同创新中心有机功能分子合成与 |
| 更多格式 | 高清、无水印(增值服务) |


{kind=link}