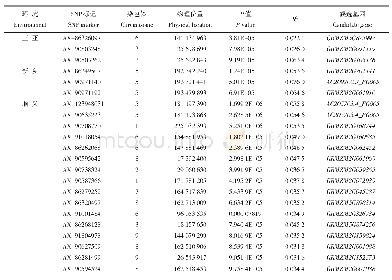
《表3 达到全基因组关联显著水平的与复杂缺血性脑卒中相关的风险位点》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
注:a染色体位置的确定参考基因组序列GRCh38/hg38;b病例组例数/对照组例数;c关联但未达到了全基因组关联显著水平。SNP为单核苷酸多态性,OR为优势比,AS为所有脑卒中,IS为缺血性脑卒中,CE为心源性脑栓塞卒中,LAA为大动脉粥样硬化性卒中,SVD为脑小血管病变,即小动脉闭塞性
绝大部分缺血性脑卒中由多个易感基因和环境因素共同引发的复杂疾病。全基因组单核苷酸多态性(single nucleotide polymorphism,SNP)数据显示,CE和LAA的遗传率相近,分别为32.6%和40.3%,而SVD的遗传率较低,仅为16.1%[41]。识别易感基因是认识复杂疾病的病因是关键所在。早期的研究基于对疾病发病机制的先验假设,仅选择一个或几个候选基因的遗传变异进行测试,结果令人失望,因为大多数被鉴定的遗传变异在独立样本中不能得到重复[42]。脑卒中最强有力的遗传关联是通过全基因组关联研究(genome-wide association study,GWAS)确定的,这种方法没有任何生物学机制的先验假设,它测试了大量的全基因组遗传变异与表型的关系[43-44]。这种方法已经识别了大量与各种性状和疾病关联的遗传变异,而且大部分结果已在独立样本中得到了令人信服的重复,有趣的是,这些识别的基因大多是之前未被怀疑过的基因,从而为潜在的生物学机制提供了新的假设[45]。大多数单个常见变异对表型的影响小,相对危险度一般仅为1.1~1.5倍[46]。考虑到遗传风险变异的影响小,要达到足够的统计功效至少需要几千以上的大样本量。因此,识别这些变异需要大型的国际联盟的创建。过去十年中,高通量基因分型技术的出现和大型国际联盟使得复杂缺血性脑卒中易感基因的发现取得了较大的进展。表3总结了业已发现的达到了全基因组关联显著水平的与缺血性脑卒中相关的遗传变异。P<5×10-8表明达到了全基因组关联显著水平,并进行了全基因组的将近一百万个独立统计检验。这些已发现的复杂缺血性脑卒中的风险基因也与房颤(PITX2和ZFHX3)、冠心病(HDAC9、chr9p21、ABO和ALDH2)、血压(HDAC9和ALDH2)、颈动脉斑块形成(MMP12)、神经炎症(TSPAN2)、平滑肌细胞和外膜细胞的发育(FOXF2)以及凝血(HABP2)相关。
| 图表编号 | XD0023601100 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2018.09.10 |
| 作者 | 农秋萍、廖小平、李其富、林蓉 |
| 绘制单位 | 海南医学院生物学教研室、海南医学院第一附属医院神经内科、海南医学院第一附属医院神经内科、海南医学院生物学教研室 |
| 更多格式 | 高清、无水印(增值服务) |


{kind=link}