《表1 不同磁序下不同位置的In掺杂相对能量变化》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
本系列图表出处文件名:随高清版一同展现
《In掺杂h-LuFeO_3光吸收及极化性能的第一性原理计算》
In在h-Lu Fe O3的掺杂位置有两种,分别记为P1和P2[21],如图1(a)所示.在包含30个原子的晶胞模型中,有4个P1位原子和2个P2位原子,首先讨论h-Lu1–x Fex O3最稳定的磁序.采用A型、C型、G型三种反铁磁结构和铁磁结构对相同的晶胞模型进行优化.为了得到最稳定的晶胞结构,以G型反铁磁序h-Lu0.833In0.167Fe O3能量为基准,比较了不同掺杂位置的In原子替换单个Lu原子对h-Lu Fe O3磁序的影响,并比较了相对总能量,结果如表1所列.结果表明,四种磁序中非线性G型反铁磁序的h-Lu0.833In0.167Fe O3能量最低,这与先前报道的计算结论一致[22],且无论是P1位掺杂还是P2位掺杂均没有改变磁序.利用(1)式计算了不同掺杂位置的掺杂形成能:
| 图表编号 | XD00211964500 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2021.02.05 |
| 作者 | 张小娅、宋佳讯、王鑫豪、王金斌、钟向丽 |
| 绘制单位 | 湘潭大学材料科学与工程学院、湘潭大学材料科学与工程学院、湘潭大学材料科学与工程学院、湘潭大学材料科学与工程学院、湘潭大学材料科学与工程学院 |
| 更多格式 | 高清、无水印(增值服务) |





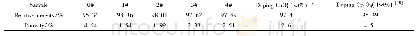
{kind=link}