《表1 路径1中各物质的键长变化表》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
nm
第一步研究了乙炔对催化剂Pd Cl2、HgCl2、MgCl2中Cl元素的夺取,并优化了共吸附在graphite(001)表面的催化剂PdCl2、HgCl2、MgCl2与乙炔.在表1中也给出了关键几何结构参数.如图2所示,催化剂Pd Cl2、HgCl2、MgCl2与乙炔吸附于CCπ键.乙炔R1中的碳碳三键与二价催化剂Pd Cl2、HgCl2、MgCl2中的Pd、Hg、Mg原子配位生成中间体M1,中间体M1中的Cl(2)原子渐渐远离R原子,并逐渐靠近C(3)原子.经过过渡态TS1逐渐生成氯乙烯基汞(中间体M2).当催化剂为PdCl2、HgCl2、MgCl2时,经过频率分析,过渡态TS1只有一个负的频率,分别是-261、-338、-257 cm-1.由表1可知,在催化剂分别为Pd Cl2、Hg Cl2、Mg Cl2时,TS1中Cl(2)—R的键长均被拉伸,而Cl(2)—C(3)的键长均被缩短.此步骤都需克服一定的能量,分别是23.9、101.7、74.1 k J/mol.Cl原子夺取到的中间体M2,吸附于变形石墨烯表面.乙炔对催化剂Pd Cl2、HgCl2、MgCl2中Cl元素的夺取具有一定的反应能量,分别为42.7、72.4、62.8 k J/mol.
| 图表编号 | XD00169384000 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2020.07.20 |
| 作者 | 覃海川、张爽、王丹阳、王薇、李来才 |
| 绘制单位 | 四川师范大学化学与材料科学学院、四川师范大学化学与材料科学学院、四川师范大学化学与材料科学学院、四川师范大学化学与材料科学学院、四川师范大学化学与材料科学学院 |
| 更多格式 | 高清、无水印(增值服务) |

![表2[HAlkyl][TFSA]S1~S5及其阴阳离子结构中N/O—H的键长与振动频率的变化值](http://bookimg.mtoou.info/tubiao/gif/SHZN202005004_03100.gif)
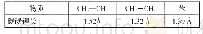



{kind=link}