《表2 Demol计算的分子W (CO) 6的振动频率》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
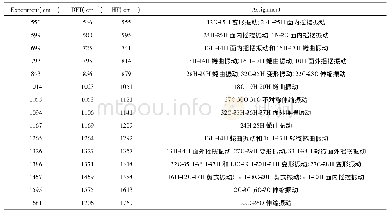
Materials Studio是一款专门的材料模拟计算软件,Dmol3是其中一个基于密度泛函理论的程序模块,具有比较丰富的泛函和比较强大的功能,可用于研究分子结构、均相催化、多相催化、分子反应性等,也可预测分子振动频率、拉曼光谱、溶解度、蒸气压、配分函数、溶解热、混合热等性质,还可用来进行周期性模型的COSMO溶剂化计算[14].缺点在于基于密度泛函理论的分子结构优化都是局域优化并非全局优化,优化后的结构及其相应的性质都与初始结构的选取是有关的.此外,不同类型的分子和材料需要选取不同的适当泛函,该程序提供的泛函仍然有待进一步丰富.我们利用Materials Studio软件中的Dmol3程序模块,在GGA-PW91水平上对W(CO)6分子进行了计算,优化后的结构见图1,W-C键的键长为0.211 4 nm、C-O键的键长为0.115 4 nm、O-W-O键角为900.图2则给出了分子的电子云、最高被占据分子轨道(HOMO)和最低未被占据分子轨道(LUMO)的电子云,13个不同的振动频率的计算结果则列于表2之中.
| 图表编号 | XD0016500000 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2018.03.01 |
| 作者 | 邝向军、贾连宝 |
| 绘制单位 | 西南科技大学理学院、西南科技大学理学院 |
| 更多格式 | 高清、无水印(增值服务) |




{kind=link}