《表7 Demol3计算获得的甲硅烷分子拉曼振动频率》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
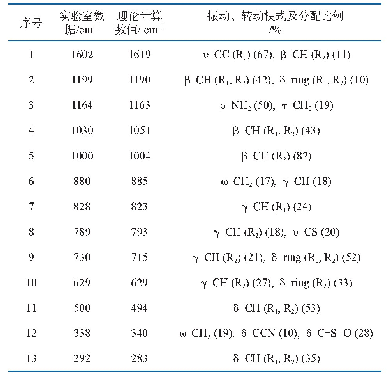
利用Materials Studio中基于密度泛函理论的Demol3程序模块,在LDA-PWC水平上,我们对磷酸根离子进行了全电子计算。图2为计算优化后获得的甲硅烷分子结构图,其中Si-H键的键长为0.149 2 nm,H-H键的键长为0.243 7 nm,H-Si-H键角为109.442°。图3给出了甲硅烷分子的最高被占据分子轨道(HOMO)和最低未被占据分子轨道(LUMO)的电子云分布。表7给出了计算得到的甲硅烷分子的拉曼振动频率。不难看出,计算获得的分子振动频率比前面推导得到的结果略小,但是吻合得还是比较好的。
| 图表编号 | XD0016261200 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2018.12.26 |
| 作者 | 邝向军 |
| 绘制单位 | 西南科技大学 |
| 更多格式 | 高清、无水印(增值服务) |


![表2 16种PAHs拉曼光谱的振动频率主要分布区域[19]](http://bookimg.mtoou.info/tubiao/gif/JGZZ202101001_02000.gif)


{kind=link}