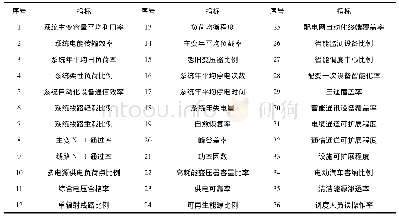
《表2 现有的Hi-C数据分析流程》
 提示:宽带有限、当前游客访问压缩模式
提示:宽带有限、当前游客访问压缩模式
Hi-C数据的分辨率是由所使用的DNA限制性内切酶等因素决定的.由于原核生物的基因组较小,在足够高的测序深度下,数据分辨率可以达到4 kb左右.但受限于实验,交互矩阵中往往带有一些系统偏差,包括与测序平台相关的偏差(如GC含量)和特定的Hi-C实验偏差(如限制性酶切位点的频率)[40].为了消除系统偏差需要对交互矩阵进行归一化处理.一类矫正方法是显性因子矫正.Yaffe和Tanay[40]通过引入一个综合的背景概率模型,并制定联合矫正程序来模拟两个区域之间的交互频率以修正交互矩阵.HiCNorm方法使用回归模型(负二项式或泊松回归)提供更快的显式校正方法同时达到与Yaffe和Tanay[40]方法相似的标准化精度[41]另一类是隐性矫正方法,这类方法没有直接讨论系统误差的来源,而是根据数据自身的特点进行矫正.SCN(sequential component normalization)是一种简单的隐性矫正归一化方法[42],该方法在移除低交互值后,分别对交互矩阵的行和列进行归一化,也就是让每行和每列的数值和为1.此外,迭代矫正(iced)是最近使用频率较高的一种方法,该方法也没有假设误差的具体来源,而是假设误差可分解,通过迭代获得最大似然解,从而消除误差[43].一些常用的Hi-C数据分析流程见表2.
| 图表编号 | XD0069551300 严禁用于非法目的 |
|---|---|
| 绘制时间 | 2019.06.20 |
| 作者 | 田六、王旭婷、华康剑、马彬广 |
| 绘制单位 | 农业生物信息湖北省重点实验室农业微生物国家重点实验室华中农业大学信息学院、农业生物信息湖北省重点实验室农业微生物国家重点实验室华中农业大学信息学院、农业生物信息湖北省重点实验室农业微生物国家重点实验室华中农业大学信息学院、农业生物信息湖北省重点实验室农业微生物国家重点实验室华中农业大学信息学院 |
| 更多格式 | 高清、无水印(增值服务) |




{kind=link}